Курсовая работа: Влияние обменных взаимодействий на вероятность дезактивации триплетных молекул акцепторов
Курсовая работа
ВЛИЯНИЕ ОБМЕННЫХ ВЗАИМОДЕЙСТВИЙ НА ВЕРОЯТНОСТЬ ДЕЗАКТИВАЦИИ ТРИПЛЕТНЫХ МОЛЕКУЛ АКЦЕПТОРОВ
Выполнил: Чекан Михаил Григорьевич
СОДЕРЖАНИЕ
Введение......................................................................................................................................... 3
ГЛАВА I. Основные закономерности сенсибилизированной фосфоресценции в твёрдых растворах органических соединений............................................................................................................ 6
Введение
С проблемой безызлучательного переноса энергии электронного возбуждения исследователям приходится сталкиваться при изучении самых разнообразных систем в таких областях науки как люминесценция, фотосинтез, радиационная физика и химия, биоэнергетика.
Фундаментальные представления о механизмах переноса энергии базируются в основном на классических результатах по фотонике синтетических органических соединений в конденсированных средах [1-4]. Хорошими модельными системами, которые часто используются для экспериментального изучения и проверки выводов теории переноса энергии триплетного возбуждения между молекулами, являются твёрдые растворы органических соединений. Это обусловлено своеобразием их физических свойств и возможностью широкого практического применения [5,6]. К таким средам относятся стекла активированные атомами или ионами, поликристаллические растворы, активированные полимерные пленки.
Основные закономерности межмолекулярного триплет-триплетного переноса энергии были установлены именно при исследовании тушения фосфоресценции молекул донора молекулами акцептора в этих системах. Однако даже для наиболее изученных донорно-акцепторных пар параметры переноса энергии триплетного возбуждения существенно отличаются у различных авторов [5-9].
Квантово – механическая теория триплет-триплетного переноса энергии в конденсированных средах была развита в работах Ферстера и Декстера [10,11].
Одним из выводов теории является то, что взаимодействие между компонентами донорно – акцепторной пары не влияет на константы скоростей как излучательной, так и безызлучательной дезактивации возбуждений акцептора. Именно это положение теории Фёрстера – Декстера (наряду с некоторыми другими) подвергается критике в новой теории переноса энергии, разрабатываемой в последнее время В.Я. Артюховым и Г.В. Майером [12-14]. Согласно этой теории взаимодействие между компонентами в донорно – акцепторной паре возмущает электронные состояния изолированных молекул еще до возбуждения молекул донора. При этом можно ожидать изменения константы скорости излучательной дезактивации энергии электронного возбуждения как в молекулах донора, так и в молекулах акцептора.
Наиболее актуальным вопрос о взаимном влиянии компонент донорно – акцепторной смеси на константы скоростей излучательной и безызлучательной дезактивации возбуждений является для межмолекулярного триплет – триплетного переноса энергии, поскольку он происходит при малых расстояниях между компонентами, так как обусловлен обменными взаимодействиями.
Таким образом, изучение механизмов дезактивации триплетных молекул в твердых растворах при их сенсибилизированном возбуждении и определение их вклада в дезактивацию возбуждений имеет актуальное значение для теории и практики межмолекулярного переноса энергии по обменно – резонансному механизму в конденсированных средах и является необходимым этапом дальнейшего развития его теоретических основ.
В связи с этим целью дипломной работы является исследование влияния взаимодействий между молекулами акцепторов в возбужденном триплетном состоянии и молекулами доноров в основном синглетном состоянии на вероятность излучательной дезактивации триплетных возбуждений акцепторов.
В соответствии с этим были поставлены следующие задачи:
- исследование спектров и кинетики сенсибилизированной фосфоресценции молекул аценафтена и нафталина, выбранных в качестве акцепторов, при возбуждении донора – бензофенона;
- рассмотрение методики определения константы скорости излучательного перехода S0 ← T акцепторов энергии;
- установление зависимости константы скорости излучательного перехода триплетных молекул акцептора от концентрации донорно-акцепторной смеси.
Глава I . Основные закономерности сенсибилизированной фосфоресценции в твёрдых растворах органических соединений.
1.1. Явление сенсибилизированной фосфоресценции и триплет-триплетный перенос энергии электронного возбуждения.
С проблемой резонансного безызлучательного переноса энергии электронного возбуждения исследователям приходится сталкиваться при изучении самых разнообразных систем в таких областях науки как люминесценция, фотосинтез, радиационная физика и радиационная химия, биоэнергетика. Этот процесс является промежуточным между актом возбуждения электронов и теми конечными процессами, где энергия возбужденных электронов используется.
Экспериментальные исследования позволили установить основные эмпирические закономерности и предложить феменологические модели описания процесса переноса энергии. Квантово-механическая теория переноса энергии в конденсированных средах была развита Т. Ферстером [1,10] для диполь-дипольного взаимодействия, и позже обобщена в работе Д. Декстера на случай мультипольных и обменных взаимодействий [11]. Дальнейшее ее развитие состояло в учете макроскопических параметров, влияющих в основном на эффективность передачи энергии. При этом основные положения теории Ферстера критическому анализу не подвергались, а изучались границы ее применимости. С начала 90-х годов В.Я. Артюховым и Г.В. Майером с сотрудниками развивается квантово-химический подход изучения переноса энергии в бихромофорных системах [15-19], результаты которого распространяются и на бимолекулярные системы. Результаты этих теоретических исследований хорошо согласуются с имеющимися экспериментальными данными. Однако механизм переноса энергии в рамках квантово-химического подхода не совпадает с механизмом, следующим из теории Ферстера.
В 1952 г. Теренин и Ермолаев наблюдали новое явление, заключающееся в том, что фосфоресценция нафталина в твердом растворе возбуждалась светом ртутной лампы с длинной волны в области 365 нм в присутствии бензофенона или бензальдегида в растворе хотя сам нафталин излучение с данной длинной волны не поглощает [20]. Теренин и Ермолаев интерпретировали указанное явление как безызлучательный перенос энергии электронного возбуждения от триплетных молекул бензальдегида или бензофенона (доноры энергии) к невозбужденным молекулам нафталина (акцепторы энергии) с переводом последних прямо в триплетное состояние, т.е. процесс протекает по схеме:
3ГД + 1ГА → 1ГД + 3ГА.
В случае явления
сенсибилизированной флуоресценции из энергетических соображений необходимо, что
бы нижний синглетный возбужденный (флуоресцентный 1Г*) уровень
донора был выше флуоресцентного уровня акцептора. Вследствие размытости
спектров поглощения органических соединений в растворах это приводит к тому,
что невозможно избирательно возбуждать молекулы донора энергии, не затрагивая
акцептор. Кроме того, вероятность безызлучательного переноса энергии между
возбужденной флуоресцентной и нормальной молекулами тем больше, чем сильнее
перекрываются спектры излучения донора энергии со спектром поглощения
акцептора, что одновременно приводит к сильной реабсорбции и вторичной
флуоресценции, которые весьма трудно учесть в конкретных условиях опыта. В
случае сенсибилизированной фосфоресценции обстоятельства значительно более
благоприятны, так как можно возбуждать донор энергии, не затрагивая акцептор,
и, кроме того, в области излучения донора энергии отсутствует сколько-нибудь
заметное поглощение акцептора. Это возможно благодаря тому, что разность
энергий между флуоресцентным и триплетным уровнем у различных классов
ароматических молекул изменяется в широком интервале значений. Потому при
облучении светом ртутных линий 3650 Å смешанного раствора бензальдегида
и нафталина будут поглощать свет и возбуждаться только молекулы бензальдегида.
Появление в спектре свечения полос фосфоресценции нафталина можно объяснить
лишь с помощью безызлучательного переноса энергии от бензальдегида в триплетном
состоянии к нафталину с переводом последнего также в триплетное состояние. Как
показали опыты, твердые растворы нафталина и других использованных акцепторов
энергии в концентрации до 0,1-0,5 М не излучают свойственных им спектров
флуоресценции и фосфоресценции при интенсивном возбуждении светом ртутных
линий 3650 Å. Схема нижних возбужденных электронных уровней молекул
донора и акцептора энергии в явлении сенсибилизированной фосфоресценции
приведена на рис.1. Слева изображены основной, флуоресцентный и фосфоресцентный
уровни донора энергии, справа – то же для акцептора энергии. Сплошными линиями
изображены электронные переходы, связанные с поглощением или излучением света,
волнистыми – переходы, при которых электронная энергия растрачивается в
тепловое движение, и, наконец, пунктирными – переходы, сопровождающие
безызлучательный перенос энергии электронного возбуждения от донора к 
|
1.2 Современные теории межмолекулярного переноса энергии в конденсированных средах.
К проблеме дезактивации возбуждений в условиях переноса энергии имеет отношение широкий круг как экспериментальных, так и теоретических вопросов. Квантово – механическая теория переноса энергии в конденсированных средах была развита Т. Фёрстером [1,2]. В ней предполагается, что перенос энергии происходит благодаря слабому диполь-дипольному взаимодействию между молекулами. И происходит он в несколько этапов:
1) сообщение энергии молекуле донора с переводом ее в возбужденное состояние;
2)
колебательная релаксация возбужденной молекулы донора до установления
теплового равновесия со средой или внутренняя конверсия в более устойчивое
возбужденное электронное состояние (для органических молекул это нижнее
возбужденное синглетное ![]() или
нижнее триплетное
или
нижнее триплетное ![]() );
);
3) непосредственная передача возбуждения от донора к акцептору;
4) колебательная релаксация в доноре до установления теплового равновесия со средой и релаксация или внутренняя конверсия в молекуле акцептора;
5) излучение или деградация энергии в акцепторе (при наличии миграции энергии может быть еще передача энергии другой такой же молекуле).
В результате процессов 4 система выходит из резонанса и обратный перенос энергии становится невозможным.
Согласно теории
возмущений [21] в квантовой механике вероятность
перехода ![]() системы из начального
состояния, описываемого волновой функцией
системы из начального
состояния, описываемого волновой функцией ![]() в
конечное
в
конечное ![]() определяется выражением:
определяется выражением:
![]() (1.1)
(1.1)
где ![]() — плотность конечных
состояний;
— плотность конечных
состояний; ![]() — оператор, инициирующий
переход (гамильтониан взаимодействия). Для приготовления начального
— оператор, инициирующий
переход (гамильтониан взаимодействия). Для приготовления начального ![]() и конечного
и конечного ![]() квантовых состояний
берутся симметризованные определенным образом произведения невозмущенных
волновых функций молекул донора и акцептора в соответствующих состояниях
квантовых состояний
берутся симметризованные определенным образом произведения невозмущенных
волновых функций молекул донора и акцептора в соответствующих состояниях ![]() ,
, ![]() . Верхние индексы 0 и 1
отвечают основному и возбужденному состояниям соответственно. В качестве
оператора перехода Ферстер берет оператор межмолекулярного взаимодействия. Это
положение теории Ферстера, а также выбор начального и конечного электронных
состояний (
. Верхние индексы 0 и 1
отвечают основному и возбужденному состояниям соответственно. В качестве
оператора перехода Ферстер берет оператор межмолекулярного взаимодействия. Это
положение теории Ферстера, а также выбор начального и конечного электронных
состояний (![]() и
и![]() ) авторы новой теории
переноса энергии (В.Я. Артюхов и Г.В Майер) считают ошибочными с позиций
современной теории электронных переходов [12,22,23].
) авторы новой теории
переноса энергии (В.Я. Артюхов и Г.В Майер) считают ошибочными с позиций
современной теории электронных переходов [12,22,23].
В адиабатическом приближении волновые функции ![]() ,
,![]() записываются через
произведение электронной волновой функции
записываются через
произведение электронной волновой функции ![]() на
колебательную
на
колебательную ![]() . Тогда в
одноэлектронном приближении, пренебрегая перекрыванием, имеем
. Тогда в
одноэлектронном приближении, пренебрегая перекрыванием, имеем
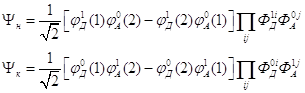
Обозначения (1) и (2) означают координаты первого и второго электронов,
а ![]() и
и ![]() – нормальные колебания в
соответствующем состоянии.
– нормальные колебания в
соответствующем состоянии.
Предполагается слабая зависимость электронного матричного элемента от координат ядер молекул (приближение Кондона), который имеет вид
![]() (1.2)
(1.2)
Выражение для вероятности (константа скорости) переноса энергии записывается в следующем виде:
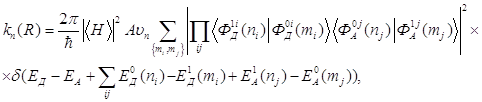 (1.3)
(1.3)
где ![]() и
и ![]() — энергии чисто электронного перехода в доноре и акцепторе
соответственно,
— энергии чисто электронного перехода в доноре и акцепторе
соответственно, ![]() означает
болцьмановское усреднение по начальному состоянию.
означает
болцьмановское усреднение по начальному состоянию.
При
конкретизации вида гамильтониана взаимодействия ![]() ,
это взаимодействие представляется в виде суммы взаимодействия внешних
электронов донора и акцептора. С учетом этого вероятность переноса энергии при
диполь-дипольном взаимодействии имеет вид:
,
это взаимодействие представляется в виде суммы взаимодействия внешних
электронов донора и акцептора. С учетом этого вероятность переноса энергии при
диполь-дипольном взаимодействии имеет вид:
 ,
(1.4)
,
(1.4)
здесь ![]() –
ориентационный фактор;
–
ориентационный фактор; ![]() и
и ![]() – квантовый выход и время
жизни возбужденного состояния донора в отсутствии тушителя;
– квантовый выход и время
жизни возбужденного состояния донора в отсутствии тушителя; ![]() – показатель преломления
среды на частоте переноса энергии;
– показатель преломления
среды на частоте переноса энергии; ![]() и
и ![]() – нормированные спектр излучения
донора и сечение поглощения акцептора соответственно.
– нормированные спектр излучения
донора и сечение поглощения акцептора соответственно.
Таким образом,
согласно теории Ферстера, в случае диполь-дипольных взаимодействий вероятность
переноса энергии ![]() пропорциональна
силам осцилляторов переходов в доноре и акцепторе, интегралу перекрытия
нормированного спектра излучения донора со спектром поглощения акцептора и
обратно пропорциональна шестой степени расстояния между молекулами.
пропорциональна
силам осцилляторов переходов в доноре и акцепторе, интегралу перекрытия
нормированного спектра излучения донора со спектром поглощения акцептора и
обратно пропорциональна шестой степени расстояния между молекулами.
Позже теория
Фёрстера была обобщена Декстером на случай мультипольных и обменных
взаимодействий [11]. Дальнейшее ее развитие состояло в учете макроскопических
параметров, влияющих в основном на константу скорости передачи энергии. При
этом считается, что взаимодействие между компонентами донорно – акцепторной
пары не влияет на константы скоростей как излучательной, так и безызлучательной
дезактивации возбуждений акцептора, поскольку для приготовления начального и
конечного квантовых состояний берутся невозмущенные волновые функции
изолированных молекул донора и акцептора энергии в соответствующих состояниях.
Именно это положение теории Фёрстера – Декстера подвергается критике в Новой
теории переноса энергии, разрабатываемой в последнее время В.Я. Артюховым и
Г.В. Майером. Перенос энергии за счет обменных взаимодействия становится
актуальным, когда кулоновская часть электронного матричного элемента
взаимодействия ![]() в (1.2)
значительно меньше обменной
в (1.2)
значительно меньше обменной
![]() . (1.5)
. (1.5)
Неравенство
(1.5) выполняется для интеркомбинационных синглет-триплетных переходов в
органических молекулах. Поэтому основной вклад в ![]() в
этом случае дает обменный интеграл. Взаимодействия такого типа названы В.Л.
Ермолаевым и А.Н. Терениным обменно-резонансными, и хотя в литературе известны
и другие термины, этот термин наиболее широко используется в настоящее время
специалистами.
в
этом случае дает обменный интеграл. Взаимодействия такого типа названы В.Л.
Ермолаевым и А.Н. Терениным обменно-резонансными, и хотя в литературе известны
и другие термины, этот термин наиболее широко используется в настоящее время
специалистами.
Рассмотрим более подробно межмолекулярный триплет-триплетный перенос энергии электронного возбуждения, происходящий по обменно-резонансному механизму.
Если
представить электронные волновые функции донора и акцептора в виде произведения
координатной волновой функции ![]() на
спиновую
на
спиновую ![]() , то обменный интеграл
, то обменный интеграл ![]() имеет вид
имеет вид
![]() . (1.6)
. (1.6)
Здесь учтено,
что ![]() описывает кулоновское
взаимодействие, которое не действует на спиновые переменные.
описывает кулоновское
взаимодействие, которое не действует на спиновые переменные.
Из (1.6)
следует, что обменный интеграл ![]() , если
, если
![]()
Возбужденное и основное состояния могут иметь разную мультипольность, т. е.
![]() (1.7)
(1.7)
Следовательно, мультипольность состояний донора и акцептора после акта передачи должна измениться одновременно.
Учитывая, что спектр излучения донора и поглощения акцептора определяются интегралами Франка-Кондона и используя (1.3) Декстер [11] записал выражение для вероятности переноса энергии по обменно-резонансному механизму в следующем виде
![]() (1.8)
(1.8)
здесь ![]() —
нормированный спектр поглощения акцептора.
—
нормированный спектр поглощения акцептора.
Поскольку
величина обменных взаимодействий пропорциональна плотности перекрывания
электронных облаков донора и акцептора энергии, которая экспоненциально убывает
с расстоянием ![]() между ними, то
параметр
между ними, то
параметр ![]() , в котором скрыта
зависимость
, в котором скрыта
зависимость ![]() от расстояния, можно
представить в виде
от расстояния, можно
представить в виде ![]() , где L — средний эффективный
боровский радиус.
, где L — средний эффективный
боровский радиус.
Таким образом, Декстер показал, что вероятность переноса
энергии ![]() по обменно -
резонансному механизму пропорциональна интегралу перекрытия спектра излучения донора
со спектром поглощения акцептора, экспоненциально убывает с увеличением
расстояния между молекулами акцептора и донора и, в отличие от
индуктивно-резонансного механизма, не зависит от сил осцилляторов переходов в
доноре и акцепторе.
по обменно -
резонансному механизму пропорциональна интегралу перекрытия спектра излучения донора
со спектром поглощения акцептора, экспоненциально убывает с увеличением
расстояния между молекулами акцептора и донора и, в отличие от
индуктивно-резонансного механизма, не зависит от сил осцилляторов переходов в
доноре и акцепторе.
Установить
непосредственную связь ![]() с
экспериментально определяемыми параметрами Декстеру не удалось. Позже в работе
[24] Инокути и Хирояма провели теоретическое рассмотрение тушения фосфоресценции
донора по обменно-резонансному механизму, основываясь на предложенной в [11]
экспоненциальной зависимости константы скорости переноса энергии от расстояния
между компонентами донорно-акцепторной пары. Обозначив
с
экспериментально определяемыми параметрами Декстеру не удалось. Позже в работе
[24] Инокути и Хирояма провели теоретическое рассмотрение тушения фосфоресценции
донора по обменно-резонансному механизму, основываясь на предложенной в [11]
экспоненциальной зависимости константы скорости переноса энергии от расстояния
между компонентами донорно-акцепторной пары. Обозначив ![]() , где
, где ![]() – критический радиус
переноса они записали выражение для
– критический радиус
переноса они записали выражение для ![]() в виде
в виде
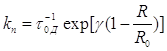 .
(1.9)
.
(1.9)
Здесь ![]() ,
где
,
где ![]() – средняя длительность
затухания донора в отсутствии акцептора.
– средняя длительность
затухания донора в отсутствии акцептора.
Обобщая основные положения и выводы теории межмолекулярного переноса энергии в конденсированных средах по обменно-резонансному механизму Ферстера-Декстера можно сказать следующее. Взаимодействие между компонентами донорно-акцепторной смеси увеличивает константу скорости безызлучательной дезактивации триплетных возбуждений в молекулах донора только за счет передачи энергии акцептору. Константы скоростей излучательной дезактивации триплетных молекул донора и триплетных молекул акцептора, а также константа скорости безызлучательной дезактивации триплетных молекул акцептора при этом должны оставаться такими же каковыми они были в однокомпонентных растворах.
Следствием этого должно быть отсутствие влияния донора на время затухания фосфоресценции акцептора и независимость квантового выхода сенсибилизированной фосфоресценции от концентрации раствора. Следует заметить, что под квантовым выходом сенсибилизированной фосфоресценции имеется ввиду, здесь и в дальнейшем, отношение числа квантов излучаемых акцептором к числу потушенных триплетных молекул донора за это же время в результате передачи энергии [25] (по определению В.Л. Ермолаева и А.Н. Теренина). Отношение же числа излученных квантов акцептором в единицу времени к числу поглощаемых квантов света донором за это же время будем называть абсолютным квантовым выходом сенсибилизированной фосфоресценции, как и в [25].
Последовательный критический анализ теории Ферстера для описания переноса энергии с позиций современной теории безызлучательных переходов был проведен В.Я. Артюховым и Г.В. Майером в [12]. Показано, что основные положения теории Ферстера ошибочны с точки зрения современной теории электронных переходов [12,13,26,27]. Так же установлены некоторые противоречия между выводами теории и экспериментальными фактами. При исследовании бихромофорных систем, содержащих ароматические молекулы, так же установлено, что величина ориентационного фактора в теории Ферстера часто не согласуется с экспериментальными данными по переносу энергии при строго определенной относительной ориентации молекул донора и акцептора.
Согласно
[12,13] волновые функции и оператор, инициирующий перенос электронной энергии,
в теории Ферстера определены неправильно. Волновые функции в выражении (1.1) и
начального и конечного состояний описывают возбужденные электронные состояния
бимолекулярной системы. Если оператор не содержит спиновых переменных, то
возможен переход только между состояниями одинаковой мультиплетности. Оператор ![]() в (1.1) по Ферстеру не
зависит от спиновых переменных и поэтому не может инициировать, согласно
[12,13], электронный переход (перенос энергии).
в (1.1) по Ферстеру не
зависит от спиновых переменных и поэтому не может инициировать, согласно
[12,13], электронный переход (перенос энергии).
Наиболее прост для
рассмотрения предложенной теории случай синглет-синглетного переноса энергии
при большом расстоянии между молекулами. При пренебрежении взаимодействием
между молекулами (в выражении для полного электронного гамильтониана
бихроморфной системы ![]() (1.10), здесь
(1.10), здесь ![]() ) для любого состояния
системы волновая функция имеет вид прямого произведения волновых функций
молекул
) для любого состояния
системы волновая функция имеет вид прямого произведения волновых функций
молекул
![]() (1.11)
(1.11)
Все состояния этой системы
соответствуют невозмущенной системе в теории возмущений. С физической точки
зрения такая ситуация соответствует полной изолированности подсистем общей
системы, т. е. набор состояний системы является просто суммой состояний
подсистем. Все свойства полной системы (в том числе и
спектрально-люминесцентные) будут аддитивны по отношению к аналогичным
свойствам подсистем. Перенос энергии электронного возбуждения в такой системе
может происходить только за счет реабсорбции излучения донора молекулой
акцептора. Здесь волновые функции молекул есть прямые произведения электронной,
колебательной и спиновой функций. Учет межмолекулярного взаимодействия (![]() ) изменяет гамильтониан
только электронной задачи, так как оператор
) изменяет гамильтониан
только электронной задачи, так как оператор ![]() не содержит взаимодействий, которые
включают неадиабатичность или смешивание спиновых состояний. С точки зрения
квантовой теории общая электронная волновая функция системы в этом случае не
может быть представлена в виде (1.11). Возникают новые состояния системы с электронными
функциями
не содержит взаимодействий, которые
включают неадиабатичность или смешивание спиновых состояний. С точки зрения
квантовой теории общая электронная волновая функция системы в этом случае не
может быть представлена в виде (1.11). Возникают новые состояния системы с электронными
функциями ![]() . Однако квантово-химические расчеты показывают, что можно
по-прежнему классифицировать состояния системы как набор возмущенных состояний подсистем:
. Однако квантово-химические расчеты показывают, что можно
по-прежнему классифицировать состояния системы как набор возмущенных состояний подсистем:
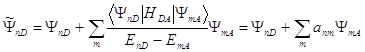 (1.12)
(1.12)
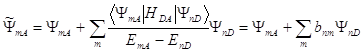 (1.13)
(1.13)
Это же показывают и
экспериментальные спектры поглощения бимолекулярных систем [12,14,15]. Обычно
их интерпретируют как почти аддитивные. Нарушение аддитивности свойств системы
существенно зависит от величин ![]() и
и ![]() т. е. от величины
межмолекулярного взаимодействия молекул D и А в соответствующих
электронных состояниях и энергетического интервала между ними. Для
осуществления процесса переноса энергии принципиально важно, что волновые
функции электронных состояний бимолекулярной системы теряют строгую
пространственную локализацию и, в общем случае, делокализованы по всей системе.
Именно пространственная делокализация электронных волновых функций состояний
компонент бимолекулярной системы является необходимым условием осуществления в
ней межмолекулярных фотофизических процессов.
т. е. от величины
межмолекулярного взаимодействия молекул D и А в соответствующих
электронных состояниях и энергетического интервала между ними. Для
осуществления процесса переноса энергии принципиально важно, что волновые
функции электронных состояний бимолекулярной системы теряют строгую
пространственную локализацию и, в общем случае, делокализованы по всей системе.
Именно пространственная делокализация электронных волновых функций состояний
компонент бимолекулярной системы является необходимым условием осуществления в
ней межмолекулярных фотофизических процессов.
Авторы [12,14,15] указывают на еще одну принципиальную ошибку исследования процесса переноса энергии согласно теории Ферстера (в том числе при классическом рассмотрении). Предполагается, что межмолекулярное взаимодействие молекул D и А включается после возбуждения системы, т. е. возбуждаются невозмущенные состояния (1.11). В действительности возбуждаются возмущенные состояния (1.12) и (1.13), а не состояния (1.11). Возмущение в виде межмолекулярного взаимодействия возникает сразу после синтезирования молекулы бихромофора или приготовления бимолекулярного раствора. Это отчетливо регистрируется в спектрах молекул. В таком подходе предлагаемая модель системы полностью соответствует исходным предположениям теории электронных переходов (в том числе процедуре приготовления возбужденного состояния), и дальнейшее исследование фотофизики бимолекулярной системы может быть проведено в рамках этой теории. Так же В.Я. Артюхов и Г.В. Майер отмечают, что в принципиальном плане любой электронный переход между электронно-колебательными состояниями системы сопровождается переносом энергии и является резонансным с точки зрения закона сохранения энергии системы.
Далее, в отличии от модели Ферстера при описании переноса энергии, здесь используется модель, рассматривающая перенос энергии, как процесс внутренней конверсии в бихромофорной или бимолекулярной системе. Для расчетов констант скоростей переходов и энергии в бимолекулярной системе применяется модель «супермолекулы» [12]. В такой модели многокомпонентная молекулярная система рассматривается как единая молекулярная система без разделения ее на отдельные фрагменты. Взаимодействие фрагментов непосредственно учитывается в гамильтониане молекулярной системы. В результате обычного квантово-химического расчета получается набор синглетных и триплетных состояний такой системы. Важно, что в состав «супермолекулы» может войти и растворитель. После расчета энергий и волновых функций электронных состояний, проводится оценка констант скоростей фотофизических процессов: константы скорости излучения, константы скоростей внутренней и интеркомбинационной конверсии. Константа скорости интеркомбинационной конверсии определяется на основе расчетов матричных элементов оператора спин-орбитального взаимодействия. Начальное и конечное состояния системы, между которыми происходит перенос энергии (внутренняя или интеркомбинационная конверсия), являются возмущенными состояниями системы. Возмущение состояний системы за счет межмолекулярного взаимодействия компонент не вызывает электронного перехода, но является необходимым условием осуществления таких переходов между состояниями, локализованными преимущественно на разных компонентах. Электронный переход инициируется традиционными для фотофизических процессов возмущениями: неадиабатичностью и спин-орбитальным взаимодействием [28].
Теорию переноса энергии основанную на квантово-химической модели В.Я. Артюхов и Г.В. Майер назвали новой теорией переноса энергии [12,23]. Авторы [12,23] указывают на следующие причины, по которым теория Ферстера удовлетворительно объясняет перенос энергии между органическими молекулами.
1)
Теория Ферстера-Декстера правильно формулирует основную зависимость
скорости переноса энергии ![]() для
обоих типов передачи энергии.
для
обоих типов передачи энергии.
2)
Зависимость вероятности переноса от разности энергетических уровней
донора энергии ![]() и акцептора
энергии
и акцептора
энергии ![]() в новой теории близка к
аналогичной зависимости
в новой теории близка к
аналогичной зависимости ![]() от
интеграла перекрывания спектров излучения молекул донора и поглощения молекул
акцептора в теории Ферстера.
от
интеграла перекрывания спектров излучения молекул донора и поглощения молекул
акцептора в теории Ферстера.
3) Правила отбора для радиационных переходов (которыми модулируется перенос энергии в теории Ферстера) и внутренней конверсии по отношению к орбитальной природе и симметрии волновых функций начального и конечного состояний системы одинаковы [14].
Поскольку
вероятность переноса энергии, согласно новой теории, зависит от разности
энергий (![]() ), то должна быть ее
зависимость от частоты перехода в пределах неоднородно уширенной полосы
), то должна быть ее
зависимость от частоты перехода в пределах неоднородно уширенной полосы ![]() перехода в акцепторе. В
теории Ферстера она менее ярко выражена.
перехода в акцепторе. В
теории Ферстера она менее ярко выражена.
Таким образом, согласно новой теории переноса энергии, можно ожидать изменения константы скорости излучательного перехода триплетных молекул акцептора в основное состояние, обусловленное взаимодействием между молекулой донора в основном состоянии и молекулой акцептора в триплетном состоянии. Так же можно ожидать зависимости константы скорости перехода молекул акцептора из основного состояния в триплетное в результате передачи им энергии от частоты перехода в пределах неоднородно уширенной 0 - 0 полосы.
1.3 Экспериментально установленные закономерности межмолекулярного триплет-триплетного переноса энергии.
Как отмечалось выше, триплет-триплетный перенос энергии был обнаружен В.Л. Ермолаевым и А.Н. Терениным в 1952 г. Они наблюдали новое явление, заключающееся в том, что фосфоресценция нафталина в твердом растворе возбуждалась светом ртутной лампы с длинной волны в области 365 нм в присутствии бензофенона или бензальдегида в растворе хотя сам нафталин излучение с данной длинной волны не поглощает.
Позднее эти явления стали известны и для жидких растворов, кристаллов и паров. И хотя триплет-триплетный перенос энергии во всех указанных случаях происходит по обменно-резонансному механизму, различные его закономерности становятся более ярко выраженными в зависимости от агрегатного состояния вещества.
Основные закономерности триплет-триплетного переноса энергии между молекулами были установлены В.Л Ермолаевым при изучении данного явления для органических соединений в твердых растворах. Эти закономерности были выявлены на основании изучения влияния акцептора на параметры фосфоресценции донора и особенностей сенсибилизированной фосфоресценции.
При экспериментальном изучении явления сенсибилизированной фосфоресценции донорно-акцепторные пары обычно выбирают таким образом, чтобы они удовлетворяли трем ниже перечисленным условиям:
1) триплетный уровень молекул акцептора расположен также или ниже соответствующего уровня молекул донора (закон сохранения энергии).
2) Первый возбужденный синглетный уровень молекул акцептора был выше соответствующего уровня молекул донора. Это позволяет возбуждать донор энергии не затрагивая при этом молекулы акцептора.
3) Время жизни триплетных молекул донора намного меньше времени жизни триплетных молекул акцептора. Выполнение этого условия позволяет отделить во времени фосфоресценцию молекул акцептора от фосфоресценции донора.
Если выполнение первого условия является необходимым для осуществления триплет-триплетного переноса энергии, то выполнение последних двух необязательно. Они необходимы лишь для удобства эксперимента.
При выполнении второго условия молекулы акцептора не будут переходить в триплетное состояние за счет поглощения возбуждающего света в случаях:
1)
если в результате взаимодействия между молекулами компонент
донорно-акцепторной смеси синглетный уровень акцептора не смещается настолько,
что он начинает поглощать возбуждающий свет в результате ![]() – перехода;
– перехода;
2)
указанное взаимодействие не изменяет вероятности излучательного перехода
молекул акцептора из триплетного состояния в основное настолько, что актуальным
при заселении их триплетного состояния становится синглет-триплетное поглощение
возбуждающего света (![]() переходы).
переходы).
В.Л. Ермолаев и А.Н. Теренин показали, что в спектрах поглощения донорно-акцепторной смеси, отсутствуют какие-либо новые полосы по сравнению с суммой спектров компонентов [14]. Спектры сенсибилизированной фосфоресценции акцепторов тождественны спектрам их фосфоресценции, возбуждаемых прямо в их полосу синглет-синглетного поглощения. Эти результаты однозначно показали, что возбуждение молекул акцептора не связано со смещением их триплетного уровня. Однако, они не могут дать однозначного ответа на вопрос влияет ли взаимодействие между компонентами донорно-акцепторной пары на положение триплетного уровня акцептора или нет. Это связано с тем, что спектры сенсибилизированной фосфоресценции акцептора широкие и на их параметры (ширину полос, положение максимума 0-0 полосы, ее форму и др.) существенно влияет как неоднородное взаимодействие молекул акцептора с раствором, так и молекул акцептора между собой. Поэтому небольшие изменения положения триплетного уровня могут маскироваться другими явлениями (например концентрационным смещением и уширением спектра и т. д.). Следовательно, для ответа на вопрос на сколько сказывается возмущение молекулами донора соответствующего электронного состояния акцептора на положение триплетного уровня последнего, необходимо создать условия, при которых смещение триплетного уровня молекул акцептора в пределах неоднородно уширенной полосы, обусловленное взаимодействиями в донорно-акцепторной паре, можно выделить и исследовать.
В.Л. Ермолаевым
и А.Н. Терениным было измерено время затухания фосфоресценции нафталина и
дифенила в этаноле при 90 К при возбуждении в собственной полосе поглощения и
при их сенсибилизированном возбуждении, когда донорами энергии являются
бензофенон и бензальдегид [1,25,29]. В пределах ошибки измерения, которая не
превышала 5% от измеряемой величины, не было обнаружено зависимости времени
жизни триплетных молекул акцептора от способа возбуждения. Это подтверждало то,
что нет такого изменения константы скорости излучательного интеркомбинационного
перехода ![]() , которое могло бы привести
к заселению триплетного состояния акцептора за счет синглет-триплетного
поглощения. Однако эти результаты не могут опровергать или подтверждать наличие
влияния взаимодействия в донорно-акцепторной паре на величину константы
скорости излучательного перехода в молекулах акцептора. Действительно,
константа скорости излучательного перехода
, которое могло бы привести
к заселению триплетного состояния акцептора за счет синглет-триплетного
поглощения. Однако эти результаты не могут опровергать или подтверждать наличие
влияния взаимодействия в донорно-акцепторной паре на величину константы
скорости излучательного перехода в молекулах акцептора. Действительно,
константа скорости излучательного перехода ![]() (рис.1)
для молекул нафталина [29] равна
(рис.1)
для молекул нафталина [29] равна ![]() . Время
жизни молекул нафталина в триплетном состоянии [29] равно
. Время
жизни молекул нафталина в триплетном состоянии [29] равно ![]() . Увеличение константы
скорости излучательного перехода в два раза (
. Увеличение константы
скорости излучательного перехода в два раза (![]() )
приведет к относительному уменьшению времени затухания менее чем на 5% (
)
приведет к относительному уменьшению времени затухания менее чем на 5% (![]() ).
).
Позднее В.Л.
Ермолаевым и его сотрудниками, получено значение для ![]() нафталина в два раза
меньшее
нафталина в два раза
меньшее ![]() [30]. В этом случае даже
увеличение константы скорости излучательной дезактивации энергии триплетного
возбуждения молекул нафталина в 4 раза повлечет за собой изменение времени
жизни их триплетных молекул менее чем на 5%. Поэтому для установления и
исследования влияния взаимодействия в донорно-акцепторной паре на вероятность
излучательного перехода молекул акцептора из триплетного состояния в основное
из кинетических экспериментов, необходимо более точное измерение параметров
триплетных молекул.
[30]. В этом случае даже
увеличение константы скорости излучательной дезактивации энергии триплетного
возбуждения молекул нафталина в 4 раза повлечет за собой изменение времени
жизни их триплетных молекул менее чем на 5%. Поэтому для установления и
исследования влияния взаимодействия в донорно-акцепторной паре на вероятность
излучательного перехода молекул акцептора из триплетного состояния в основное
из кинетических экспериментов, необходимо более точное измерение параметров
триплетных молекул.
Количественные измерения некоторых характеристик явления сенсибилизированной фосфоресценции, выполненные Ермолаевым и Терениным показали следующее.
1). Характер зависимости интенсивности
сенсибилизированной фосфоресценции акцептора (![]() )
от концентрации донора подчиняется следующей закономерности
)
от концентрации донора подчиняется следующей закономерности
![]() ,
(1.14)
,
(1.14)
где ![]() –
концентрация донора,
–
концентрация донора, ![]() – молярный
десятичный коэффициент поглощения донора для длинны волны возбуждающего света,
– молярный
десятичный коэффициент поглощения донора для длинны волны возбуждающего света, ![]() – толщина кюветы. Вероятно
это обусловлено тем, что интенсивность сенсибилизированной фосфоресценции
пропорциональна количеству квантов, поглощаемых молекулами донора.
– толщина кюветы. Вероятно
это обусловлено тем, что интенсивность сенсибилизированной фосфоресценции
пропорциональна количеству квантов, поглощаемых молекулами донора.
2). Зависимость интенсивности сенсибилизированной фосфоресценции от концентрации акцептора имеет также экспоненциальный характер при фиксированной концентрации донора и описывается подобной формулой
![]() ,
(1.15)
,
(1.15)
где ![]() –
постоянная величина;
–
постоянная величина; ![]() – концентрация
акцептора энергии;
– концентрация
акцептора энергии; ![]() .
.
Наличие закономерностей (1.14) и (1.15) не противоречит возможности изменения вероятностей излучательной и безызлучательной дезактивации триплетных молекул акцептора при добавлении в раствор молекул донора.
Возможно, чтобы разобраться в деталях изменения
интенсивности сенсибилизированной фосфоресценции ![]() ,
необходимо установить причину изменения числа квантов излучаемых
сенсибилизированной фосфоресценцией. Для интенсивности сенсибилизированной
фосфоресценции можно записать
,
необходимо установить причину изменения числа квантов излучаемых
сенсибилизированной фосфоресценцией. Для интенсивности сенсибилизированной
фосфоресценции можно записать
![]() ,
(1.16)
,
(1.16)
здесь ![]() –
концентрация триплетных молекул акцептора;
–
концентрация триплетных молекул акцептора; ![]() –общая
концентрация молекул акцептора, участвующих в излучении сенсибилизированной
фосфоресценции;
–общая
концентрация молекул акцептора, участвующих в излучении сенсибилизированной
фосфоресценции; ![]() – относительная
заселенность триплетного уровня, показывающая какая часть молекул акцептора от
– относительная
заселенность триплетного уровня, показывающая какая часть молекул акцептора от ![]() находится в триплетном
состоянии.
находится в триплетном
состоянии.
Как видно из (1.16), изменение любой из трех величин ![]() ,
, ![]() , и
, и ![]() – может привести к
изменению
– может привести к
изменению ![]() . Работы по установлению и
определению вклада каждой из этих величин в изменение интенсивности
сенсибилизированной фосфоресценции в литературе отсутствуют. Так же как и
отсутствовали на момент начала нашего исследования методики определения
. Работы по установлению и
определению вклада каждой из этих величин в изменение интенсивности
сенсибилизированной фосфоресценции в литературе отсутствуют. Так же как и
отсутствовали на момент начала нашего исследования методики определения ![]() и
и ![]() при сенсибилизированном
заселении триплетного уровня молекул.
при сенсибилизированном
заселении триплетного уровня молекул.
Важнейшими параметрами фотопроцессов, значение которых
непосредственно зависит от путей деградации энергии электронного возбуждения,
являются их квантовые выходы. Квантовый выход сенсибилизированной
фосфоресценции, определенный Ермолаевым и Терениным как отношение числа квантов
сенсибилизированной фосфоресценции, испущенных акцептором, к числу потушенных
квантов фосфоресценции донора [20,29], согласно выводам теории Ферстера, не
должен завесить от концентрации раствора. Экспериментально концентрационная
зависимость квантового выхода сенсибилизированной фосфоресценции была исследована
Ермолаевым. Было показано, что при изменении концентрации акцептора в пределах
от ![]() до
до ![]() моль/л значение квантового
выхода сенсибилизированной фосфоресценции в пределах ошибки эксперимента не
изменялось. Погрешность измерений квантового выхода при этом составила 15-20%.
моль/л значение квантового
выхода сенсибилизированной фосфоресценции в пределах ошибки эксперимента не
изменялось. Погрешность измерений квантового выхода при этом составила 15-20%.
На наш взгляд, эти результаты не могут однозначно опровергать возникновение дополнительных каналов дезактивации триплетных молекул акцептора энергии в присутствии донора в сравнении с однокомпонентным раствором. Действительно, если происходит смешение триплетных состояний донора и акцептора энергии, которые удовлетворяют вышеперечисленным условиям, то можно ожидать увеличения константы скорости излучательного перехода в молекулах акцептора. Поскольку вклад состояния донора будет тем больше, чем меньше расстояние между компонентами бимолекулярной системы, то с увеличением концентрации раствора вероятность излучательного перехода будет возрастать. Это, в свою очередь, повлечет рост квантового выхода сенсибилизированной фосфоресценции с ростом концентрации раствора.
С другой стороны, при увеличении концентрации раствора возрастает вероятность образования гетероассоциатов, которые эффективно тушат триплетные состояния акцептора. Поскольку этот вид тушения усиливается с увеличением концентрации раствора, то следствием его будет концентрационное падение квантового выхода сенсибилизированной фосфоресценции. При определенных условиях эти два механизма могут компенсировать влияние друг друга на концентрационную зависимость квантового выхода сенсибилизированной фосфоресценции. Однако этот вопрос оставался не изученным.
В стеклообразных растворах при 77 К спектры фосфоресценции акцептора имеют диффузный характер как при прямом, в отсутствие донора, так и при сенсибилизированном возбуждении и заметного различия между ними не наблюдается. Поэтому извлечь какую – либо информацию об особенностях взаимодействия партнеров в донорно – акцепторной паре из спектров фосфоресценции достаточно сложно. По видимому, это и является причиной того, что их изучению посвящено сравнительно малое число работ, имеющихся в литературе.
Новые возможности для спектральных исследований переноса энергии дает открытый в 1952 г. Э.В. Шпольским, А.А. Ильиной и Л.А. Климовой эффект резкого сужения спектральных полос люминесценции ряда ароматических углеводородов в замороженных н.- парафиновых растворах [31]. Попытки получить квазилинейчатый спектр [32-36] сенсибилизированной фосфоресценции не дали положительного результата. Тонкая структура спектра излучения акцептора размывалась при переходе к сенсибилизированному возбуждению. Квазилинейчатые спектры сенсибилизированной фосфоресценции удавалось получить лишь в том растворителе, в котором и акцептор и донор имеют каждый в отдельности при выбранной концентрации квазилинейчатые спектры [37-40]. Было установлено, что эффективность образования донорно – акцепторных пар в этих условиях различна для различных центров. Это проявляется в отличии мультиплетной структуры спектров при прямом, в отсутствие донора, и сенсибилизированном возбуждении, что объясняется образованием нескольких излучающих и поглощающих центров с разной эффективностью передачи энергии. Причина различной эффективности переноса энергии связывается с зависимостью обменно – резонансного взаимодействия от взаимной ориентации партнеров в матрице растворителя. Так же были изучены спектры сенсибилизированной фосфоресценции хинолина и нафталина в матрицах н.- парафинов от пентана до октана при 77 К [41]. Из сопоставления мультиплетов обычной и сенсибилизированной фосфоресценции сделан вывод, что они различаются как по числу компонентов, так и по положению и относительной интенсивности. Было выдвинуто предположение, что мультиплетность в спектре акцептора при сенсибилизированном возбуждении и его квазилинейчатая структура обусловлены эффектом селекции в переносе энергии. Этот эффект селекции может быть связан как с особенностями взаимного расположения энергетических уровней донора и акцептора, так и с особенностями взаимного расположения партнеров в донорно – акцепторной паре. Эту гипотезу авторы [41] подтверждают различием мультиплетной структуры спектров сенсибилизированной фосфоресценции акцептора в одном и том же растворителе в случае различных доноров. Однако возможна и иная интерпретация результатов этой работы. Не исключено, что за квазилинейчатые спектры, ответственны молекулы акцептора, находящиеся в агрегатах донора. Так в некоторых работах [42,43] наблюдался квазилинейчатый спектр сенсибилизированной фосфоресценции нафталина в кристаллах бензофенона при возбуждении через основу. И было установлено, что триплет – триплетный перенос энергии эффективно осуществляется, если молекулы акцептора внедрены в агрегаты донора.
Следует отметить, что даже для наиболее структурных спектров квазилинии сенсибилизированной фосфоресценции уширены в сравнении с квазилиниями обычной фосфоресценции в тех же условиях [37,38]. Связано ли это уширение только с влиянием донора на формирование микроматрицы или же здесь проявляется непосредственное влияние донора на параметры фосфоресценции акцептора – дать однозначный ответ на этот вопрос, на основании экспериментального материала имеющегося к настоящему времени, не представляется возможным.
1.4 Выводы к первой главе.
Анализ литературы по триплет-триплетному переносу энергии между примесными молекулами в конденсированных средах позволяет сделать следующие выводы. Проблема безызлучательного переноса энергии электронного возбуждения в жидких и твердых телах является одной из фундаментальных проблем физики конденсированного состояния и широко исследуется в течение многих десятилетий. Первый период после открытия Терениным и Ермолаевым межмолекулярного триплет-триплетного переноса энергии характеризуется в основном установлением основных закономерностей этого явления и сопоставлением их с выводами теории Ферстера-Дектстера передачи энергии по обменно-резонансному механизму. Некоторые несоответствия между ними удается устранить уточнением указанной теории не затрагивая ее основ. Однако ряд важных вопросов, связанных с самой природой переноса энергии, остаются открытыми. В частности остаются без ответа такие вопросы: насколько сильно взаимодействие в донорно-акцепторной паре возмущает электронные состояния компонентов, как это возмущение влияет на константы скоростей дезактивации их триплетных возбуждений? Насколько изменение констант скоростей переходов в результате таких взаимодействий сказывается на квантовом выходе и кинетике сенсибилизированной фосфоресценции? Необходимость решения этих вопросов для дальнейшего развития данной области фотофизики конденсированных сред и практического ее применения не вызывает сомнения. Эти вопросы, в общем случае не решены и в новой теории переноса энергии, развиваемой в последнее десятилетие Артюховым и Майером. Хотя одним из основных ее положений является то, что взаимодействие между компонентами в донорно-акцепторной паре возмущает соответствующие электронные состояния компонентов еще до возбуждения молекул донора. Однако, в отличии от теории Ферстера-Декстера, она не исключает влияния этих взаимодействий на вероятность дезактивации триплетных возбуждений.
Глава II. Методика экспериментальных исследований.
2.1 Растворители и соединения.
Важнейшим источником информации о строении и свойствах молекул и твердых тел являются их оптические спектры [3,44,45]. Для решения поставленных задач особый интерес представляют электронные спектры, поскольку именно в них наиболее отчетливо проявляется связь оптических свойств молекулы (или кристалла) с химическими, фотофизическими и фотохимическими свойствами. Но наиболее важным для нас является то, что электронные спектры оказываются наиболее чувствительными к различного рода внутри- и межмолекулярным взаимодействиям и служат ценным средством исследования взаимодействия молекул между собой и с окружением [2,21,24,46]. Поэтому метод оптической спектроскопии был выбран в качестве одного из основных методов исследования.
В экспериментальных исследованиях триплетных молекул важное место, наряду со спектральными, занимают кинетические методы [1,2,47], то есть изучение процессов заселения и распада возбужденных состояний. Определенные из кинетических экспериментов параметры являются характеристиками, как самих молекул, так и их взаимодействия между собой и с матрицей, в случае примесных центров. Особенно важным является то, что параметры кинетики (время накопления и время дезактивации возбужденных состояний), определяются константами скоростей соответствующих переходов и, следовательно, позволяют извлечь информацию, о путях дезактивации триплетно возбужденных молекул. Этим обусловлена необходимость использования кинетических методов для установления и изучения механизмов дезактивации триплетных состояний органических молекул в твердых матрицах при их сенсибилизированном возбуждении.
2.1.1. Растворители.
В работе исследовались стеклообразные растворы донорно-акцепторных смесей. В стеклах примесные центры распределены по объему образца равномерно, что позволяет исследовать зависимость люминесцентных характеристик как от среднего расстояния между молекулами различных компонент смеси, так и от расстояния между молекулами каждой из компонент в отдельности. В качестве растворителей, замерзающих в виде стекла при быстром охлаждении до 77 К, были выбраны этанол и толуол. Эти растворители широко используются при исследовании триплет-триплетного переноса энергии электронного возбуждения между примесными молекулами при 77К в качестве матриц [1], что позволяло сравнивать измеренные параметры люминесценции с имеющимися в литературе данными.
Этанол дополнительно очищался путем двухкратной перегонки. Обезвоживание его при этом не производилось. Критерием его чистоты являлось отсутствие люминесценции при 77 К.
Толуол использовался марки «ХЧ» или «для спектроскопии». И в том и другом случае он подвергался дополнительной очистке путем однократной перегонки. Критерием его чистоты также служило отсутствие люминесценции при 77 К.
Известно, что толуол при охлаждении либо стеклуется, либо кристаллизуется. Характер его отвердевания определяется скоростью замораживания. Поэтому, прежде всего, были изучены условия, при которых толуол замерзал в виде стекла. Было установлено, что в кювете с толщиной стенок 0,5мм и диаметром 2 мм, толуол всегда стеклуется при быстром погружении его в азот, если не давать образовываться тепловой «рубашке» вокруг кюветы. Последнего условия можно добиться перемещением кюветы в жидком азоте до момента времени, когда температура растворителя станет меньше 100 К. Если же нагревать толуол, затвердевший в виде стекла, от 77К, то при температуре Т=133 К наступает фазовый переход стекло-кристалл. Выбор толуола в качестве основного растворителя, замерзающего в виде стекла, обусловлен высокой растворимостью в нем органических соединений, в том числе и используемых в качестве донорно-акцепторных пар.
2.1.2. Донор энергии.
С учетом требований, предъявляемых к донорно-акцепторным парам в качестве донора энергии был выбран бензофенон. Квантовый выход триплетных состояний бензофенона близок к единице [48]. Фосфоресценция бензофенона в матрицах при низких температурах достаточно хорошо изучена. Основные характеристики донора энергии приведены в таблице 2.1.
Таблица 2.1.
Основные характеристики бензофенона.
| Соединение | Растворитель |
Т1-уровень, см –1 |
S1-уровень, см –1 |
τ-фосфор., с |
Источники |
| Бензофенон |
Этанол (90 К) |
24250 | 26000 |
4,7ּ10 -3 |
[59] |
|
Этанол (77 К) |
–– | –– |
6,2ּ10 -3 |
[87] | |
|
Этанол+эфир (2:1; 77 К) |
–– | –– |
5,4ּ10 -3 |
[87] | |
|
Этанол+метил- циклогексан (2:1; 77 К) |
–– | –– |
5,0ּ10 -3 |
[87] |
Бензофенон. Люминесцентные и спектральные характеристики бензофенона изучались многими авторами как в конденсированной среде для твердой [49-53] и жидкой фазы [54], так и для молекул в растворах [20,40,49,56] и парах [48,55]. Исследование спектров фосфоресценции при 90К кристаллического и стеклообразного бензофенона, а также его раствора в спиртово-эфирной смеси [49] показало, что структура спектра и распределения интенсивности в нем одинаковы во всех трех случаях. Однако положение максимумов полос их ширина и время затухания фосфоресценции в каждом состоянии были различны, что дает возможность использовать эти параметры для идентификации центров являющихся донорами энергии. Это весьма актуально в данной работе, поскольку бензофенон использовался нами как основной донор энергии. Ниже в таблице 2.2 приведены характеристики фосфоресценции бензофенона в различных его состояниях, взятые из работы [49].
Таблица 2.2.
Характеристики фосфоресценции бензофенона при 90 К в различных его состояниях [49].
| Состояние | Положение максимума 0-0 полосы, нм |
Полуширина 0-0 полосы, см –1 |
Время затухания фосфоресценции, с |
|
Раствор (спирт-эфир) |
414 | 700 |
4,7ּ10 -3 |
| Кристаллический | 416 | 300-600 |
≈ 7ּ10 -4 |
| Стеклообразный | 427 | 700 |
3,4ּ10 -3 |
Бензофенон [50,54] может существовать в трех твердых модификациях (фазах): стабильной кристаллической (α), нестабильной кристаллической (β) и стеклообразной (х). Спектр фосфоресценции монокристаллов α-модификации и спектр фосфоресценции х-модификации бензофенона [51] в температурном интервале от 77 к до 200 К состоит из одной серии широких полос с характерными для карбоксильной группы С ═ О колебательным интервалом 1640 см –1. Такой же вид имеет спектр растворов [49] бензофенона при 77 К.
Характерную температурную зависимость испытывают квантовый выход и время затухания фосфоресценции α-модификации [51]. При понижении температуры от 200 до 30 К относительный квантовый выход фосфоресценции увеличивается в 2 раза. Аналогичную зависимость в указанном температурном интервале испытывает и время затухания фосфоресценции.
В отличие от β-модификации, квантовый выход х-модификации [51] в интервале от 220 до 4,2 К увеличивается на два порядка. Время затухания фосфоресценции при этом меняется от 10 –4 с при 200 К до 4,8·10 –3 с при 4,2К. Важно отметить, что аморфные пленки бензофенона имеют такой же характер температурной зависимости квантового выхода и времени затухания фосфоресценции, как и образцы х-модификации. Эти результаты необходимо учитывать при проведении температурных и других измерений в концентрированных н.-парафиновых растворах, поскольку в них высока вероятность образования молекулами бензофенона, вытесненными на поверхность кристаллов растворителя, аморфных пленок.
Характерную
зависимость от температуры [51] испытывает и положение спектра фосфоресценции
х-модификации. Так, при понижении температуры от 220 до 97 К спектр смещается в
длинноволновую область на ![]() . При
дальнейшем понижении температуры до 4,2 К максимум смещается в противоположную,
коротковолновую область так же на
. При
дальнейшем понижении температуры до 4,2 К максимум смещается в противоположную,
коротковолновую область так же на ![]() . В
результате при температурах 220 и 4,2 К положение спектров в шкале частот
совпадает.
. В
результате при температурах 220 и 4,2 К положение спектров в шкале частот
совпадает.
Спектр фосфоресценции β-модификации бензофенона [51] состоит из двух серий. Относительная интенсивность спектров этих серий существенно зависит от температуры. Фосфоресценция центров, ответственных за эти спектры, также заметно отличается. Так, при 77 К времена затухания их фосфоресценции равны 0,11·10 –3 и 2,7·10 –3 с. Эти особенности фосфоресценции β-модификации бензофенона позволяют ее легко идентифицировать и отличить от фосфоресценции одиночных молекул.
Авторами [56] исследована кинетика затухания фосфоресценции бензофенона в различных растворителях при 77 К. При этом были получены следующие значения времени затухания: в этаноле 2 мс, в смеси этанол-эфир (2:1) 5,4 мс, в смеси эфир-метилциклогексан (2:1) 5,0 мс. Ошибка измерений составляла ± 0,2 мс.
Таким образом, люминесцентные характеристики фосфоресценции бензофенона испытывают существенные изменения в зависимости от его агрегатного состояния, от растворителя и условий ее наблюдения. Это дает возможность идентифицировать центры излучения, которые образуются при замораживании концентрированных растворов бензофенона.
2.1.3 Акцепторы энергии.
В качестве акцепторов энергии использовались нафталин, аценафтен. Люминесцентные характеристики этих соединений в матрицах хорошо известны в литературе и приведены в таблице 2.3 со ссылкой на источник. Однако некоторые из них необходимо было уточнить или изучить влияние на них различных факторов (температуры, концентрации, растворителя и т. д.). Необходимость проведения таких исследований обусловлена тем, что методика определения механизмов дезактивации триплетных молекул акцептора при их сенсибилизированном возбуждении включала исследования влияния донора на люминесцентные характеристики молекул акцептора. Чтобы выделить в чистом виде эти изменения люминесцентных характеристик нами были проведены вышеуказанные исследования. Поэтому в таблице 2.3 наряду с литературными данными об основных люминесцентных характеристиках акцепторов, также приведены результаты измерений. Ниже приведены результаты таких исследований для каждого соединения, где также описаны необходимые литературные данные о них, которые не включены в таблицу.
Как видно из таблицы 2.2 и таблицы 2.3 для любой комбинации выбранных донора и акцепторов энергии выполняются требования к донорно-акцепторным парам [1]. А именно:
1. Триплетный уровень всех молекул донора расположен выше соответствующего уровня любой из молекул акцептора, что делает возможным перенос энергии по закону сохранения энергии.
2. Флуоресцентный уровень всех молекул, используемых в качестве донора, лежит ниже соответствующего уровня любой молекулы выбранной в качестве акцептора. Это позволило избирательно возбуждать только донор, не затрагивая молекулы акцептора. При выполнении этого условия исключается также синглет-синглетный перенос энергии из-за неблагоприятного расположения энергетических уровней.
Таблица 2.3.
Основные характеристики акцепторов энергии.
| Соединение | Растворитель |
Т1-уровень, см –1 |
S1-уровень, см –1 |
τфос, с |
Источники |
| Нафталин | Смесь этанола и диэтилового эфира | 21250 | 31750 | 2,3 | [29] |
| Толуол | –– | –– | 2,40 | Настоящая работа | |
| Аценафтен | Этанол | –– | –– | 2,95 | Настоящая работа |
| толуол | –– | –– | 2,96 | Настоящая работа |
3. Время затухания всех молекул акцепторов в замороженных растворах при 77 К составляет несколько секунд, что на два порядка больше времени затухания фосфоресценции доноров. Благодаря этому, после прекращения возбуждения уже спустя 0,1 с свечение полностью определяется фосфоресценцией акцептора.
Нафталин. Нафталин является одним из наиболее изученных органических соединений, с точки зрения спектральных и люминесцентных характеристик, и широко используется в качестве объекта исследования при изучении фотофизических процессов в молекулах [44,57-59] в том числе и при изучении триплет-триплетного переноса энергии [25]. Именно с использованием его в качестве акцептора энергии было открыто [20], как уже отмечалось, явление триплет-триплетного переноса энергии электронного возбуждения.
Параметры
триплетного состояния нафталина в стеклообразных матрицах определялись многими
авторами, в том числе и в [1,20]. Константа скорости излучательного перехода
согласно [1] равна ![]() , по данным
работы [30] она равна
, по данным
работы [30] она равна ![]() с-1.
Время затухания фосфоресценции согласно [1] было 2,3 с, в [30] оно равнялось
2,50 с. Определенное нами время затухания фосфоресценции нафталина в
стеклообразном толуоле при концентрации раствора СН= 0,05 моль/л
было
с-1.
Время затухания фосфоресценции согласно [1] было 2,3 с, в [30] оно равнялось
2,50 с. Определенное нами время затухания фосфоресценции нафталина в
стеклообразном толуоле при концентрации раствора СН= 0,05 моль/л
было ![]() с. С увеличением
концентрации раствора, начиная с 0,1 моль/л, скорость затухания увеличивается.
Причиной этому является концентрационное тушение.
с. С увеличением
концентрации раствора, начиная с 0,1 моль/л, скорость затухания увеличивается.
Причиной этому является концентрационное тушение.
Времена затухания фосфоресценции нафталина в различных растворителях для случая, когда влиянием реабсорбции и концентрационным тушением можно пренебречь, приведены в таблице 2.5.
Поскольку при исследовании триплет-триплетного переноса энергии используются высокие концентрации растворов, то актуальной является проблема образования агрегатов различной степени сложности и знание их люминесцентных и спектральных характеристик. Шпольский с сотрудниками [60] показали, что спектры флуоресценции растворов нафталина в н.-гептане, состоящие при малых концентрациях из сравнительно широких молекулярных полос, приобретают при увеличении концентрации свыше 10 –2 моль/л квазилинейчатый характер. Объяснение этого факта построено на предположении, что за спектр флуоресценции, представленный широкими молекулярными полосами ответственны молекулы, «неустроенные» в кристаллической матрице растворителя. Повышение концентрации раствора приводит к агрегации таких молекул. Агрегаты нафталина характеризуются своим собственным спектром поглощения и не люминесцируют. Поэтому при агрегации «неустроенных» молекул, число «устроившихся» молекул в матрице увеличивается относительно «неустроенных» и становится заметным квазилинейчатый спектр. Следует заметить, что наличие агрегатов может существенно изменять люминесцентные характеристики раствора при наличии миграционно-ускоренного тушения [59,61]. Необходимо также учитывать то, что дезактивация возбужденных состояний нафталина может эффективно происходить [62-64] через образование эксимеров и эксиплексов.
Аценафтен. Спектральные и люминесцентные характеристики аценафтена в замороженных растворах при 77 К близки к подобным характеристикам нафталина (таблица 2.3). Аценафтен имеет высокую растворимость в толуоле, использованного нами в качестве основного растворителя. Это позволяло исследовать перенос энергии триплетного возбуждения с его участием в качестве акцептора при малых межмолекулярных расстояниях.
Затухание
фосфоресценции аценафтена, по результатам наших исследований, в стеклообразном
толуоле при 77 К в интервале концентраций от 0,01 до 0,1 моль/л было
экспоненциальным с характерным временем ![]() =2,96
с. Увеличение концентрации свыше 0,1 моль/л приводило к падению времени
затухания. Как и в случае с нафталином, это было связано с концентрационным
тушением.
=2,96
с. Увеличение концентрации свыше 0,1 моль/л приводило к падению времени
затухания. Как и в случае с нафталином, это было связано с концентрационным
тушением.
Эти значения времени затухания фосфоресценции аценафтена, полученные в растворителе, представлены в таблице 2.3.
2.2 Схема экспериментальной установки и методика получения спектров и измерения параметров кинетики.
Экспериментальные исследования были выполнены на установке, блок схема которой приведена на рис. 2.1. Она позволяла получать и исследовать спектры поглощения и люминесценции, кривые разгорания и затухания фосфоресценции, а также зависимости люминесцентных характеристик изучаемых объектов от температуры.
Экспериментальная установка была собрана на базе монохроматора СДМС с дифракционной решеткой 1200 шт/мм, работающей в первом порядке. Обратная линейная дисперсия равнялась 1,2 нм/мм. Данная решетка позволяла исследовать спектр в диапазоне длин волн от 250 до 700 нм. С помощью монохроматора можно было выделять для исследования вибронные полосы в спектре фосфоресценции молекул, узкие спектральные участки в полосах, а также исследовать суммарную интенсивность свечения без разложения в спектр при работе решетки в нулевом порядке. В некоторых опытах, при работе решетки в нулевом порядке, использовалась комбинация различных фильтров для выделения широкого участка спектра в нужной его области. Блок поворота решетки 2 включал в себя синхронный двигатель СД-54 с редуктором, позволяющим изменять скорость ее вращения в широких пределах. Градуировка монохроматора проверялась по линиям излучения ртутной лампы низкого давления. Исследуемый образец 3 помещался в сосуд Дьюара 4 с жидким азотом, который был расположен в темновой камере 5.
Доноры
возбуждались излучением ртутной лампы 6 типа ДРТ – 230 с фильтрами выделяющими
линию 365 нм или азотным лазером 7 типа ЛГИ – 21 (![]() нм)
с частотой следования импульсов 100 Гц. Плотность мощности в импульсе для
нерасфокусированного луча лазера составляла примерно 10 4 Вт/см
2. Поскольку в таких условиях не исключались двухфотонные процессы, то
для контроля проводились опыты при уменьшенной с помощью нейтральных фильтров
(металлических сеток) мощности в 10 раз. В некоторых опытах в качестве
источника сплошного спектра использовалась ксеноновая лампа ДКСШ-150 с
фильтрами выделяющими нужную спектральную область.
нм)
с частотой следования импульсов 100 Гц. Плотность мощности в импульсе для
нерасфокусированного луча лазера составляла примерно 10 4 Вт/см
2. Поскольку в таких условиях не исключались двухфотонные процессы, то
для контроля проводились опыты при уменьшенной с помощью нейтральных фильтров
(металлических сеток) мощности в 10 раз. В некоторых опытах в качестве
источника сплошного спектра использовалась ксеноновая лампа ДКСШ-150 с
фильтрами выделяющими нужную спектральную область.
При изучении
обычной фосфоресценции акцепторов, люминесценция возбуждалась светом ртутной
лампы в области 313 и 290 нм.
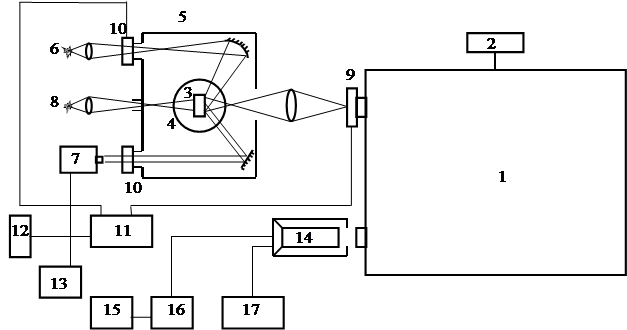 Рис.
2.1. Спектрофлуориметрическая установка для спектральных и кинетических
измерений.
Рис.
2.1. Спектрофлуориметрическая установка для спектральных и кинетических
измерений.
1. Монохроматор СДМС
2. Блок поворота решетки
3. Исследуемый образец
4. Сосуд Дьюара
5. Темновая камера
6. Лампа ДРТ – 230 (или ДКсШ – 150)
7. Азотный лазер типа ЛГИ-21
8. Дейтериевая лампа ДДС-3
9. Электромеханические затворы
10. Электромеханические затворы
11. Реле времени
12. Переносной пульт управления
13. Калибратор импульсных напряжений типа В 1-5
14. Фотоэлектронный умножитель типа ФЭУ-38
15. Двухкоординатный самописец типа Н-307
16. Источник питания фотоэлектронного умножителя
17. Катодный повторитель
Для отделения сенсибилизированной фосфоресценции акцептора от фосфоресценции донора и изучения закона затухания фосфоресценции на различных ее стадиях использовались электромеханические затворы 9 и 10, управляемые с помощью электронных реле времени 11, с применением переносного пульта управления 12. Время срабатывания затворов (перекрывания светового потока) не превышало 5 мс. Электронные реле времени позволяли изменять дискретно задержку времени между началом регистрации и прекращением возбуждения от 0,1 до 30 с. Это давало возможность отделять во времени фосфоресценцию акцептора от фосфоресценции донора в области перекрывания их спектров, даже если интенсивность фосфоресценции донора значительно превышала интенсивность фосфоресценции акцептора. Это также позволяло исследовать кинетику затухания фосфоресценции на различных ее стадиях. Система управления затворами давала возможность формировать световые импульсы возбуждения различной длительности, что было необходимо для изучения зависимости кинетики затухания от продолжительности возбуждения.
Регистрирующая часть установки включала в себя фотоэлектронный умножитель 14 типа ФЭУ-38 и двух координатный самописец 15 типа Н-307 (или запоминающий осциллограф с электронной памятью С 8-13).
В качестве источника питания 16 фотоэлектронного умножителя использовался высоковольтный стабилизированный источник высокого напряжения ВС-2С. Для согласования низкого входного сопротивления самописца и высокого выходного сопротивления фотоэлектронного умножителя использовался катодный повторитель 17, постоянную времени которого можно было изменять и устанавливать одну из следующих величин: 0.01, 0.02, 0.05, 0.1, 1.0, и 2.0 секунды. Для уменьшения случайных шумов при записи спектров, значение постоянной времени было 0.1 с, 1.0 с или 2.0 с и зависело от скорости записи. Кинетические кривые записывались при постоянной времени 0.01 с. Линейность работы усилителя постоянного тока проверялась при помощи калиброванных нейтральных фильтров. Цена деления блока временной развертки самописца проверялась с помощью секундомера выверенного по сигналам точного времени в течение суток. Механическая постоянная времени самописца не превышала 0.03 с. В случае, когда сигнал регистрировался осциллографом, катодный повторитель не использовался.
Величина погрешности при определении времени разгорания и затухания фосфоресценции в секундном диапазоне обуславливалась флуктуациями фототока, нелинейностью усилителя, погрешностью блока временной развертки и механической постоянной самописца. Три последних источника по данным многократных проверок могли дать в сумме систематическую ошибку не более 1%. Для уменьшения влияния флуктуаций фототока измерения повторялись 5-10 раз и случайная ошибка в каждом конкретном случае находилась с использованием коэффициентов Стьюдента [65] при доверительной вероятности 0,90.
При определении относительной заселенности триплетного уровня молекул и константы скорости перехода молекул акцептора из основного состояния в триплетное основной вклад в ошибку вносит случайная ошибка, возникающая при измерении времени разгорания и затухания фосфоресценции. Определенная, с учетом сказанного, абсолютная ошибка при измерении относительной заселенности триплетного уровня молекул акцептора равнялась 0,02 единицы, а для константы скорости перехода молекул акцептора в триплетное состояние 0,01 с –1.
Была исследована кинетика затухания сенсибилизированной фосфоресценции для различных донорно – акцепторных пар в стеклообразных растворителях при 77 К и произведено ее сравнение с затуханием обычной фосфоресценции, в отсутствие донора, при одних и тех же концентрациях акцептора.
2.3 Методика определения константы скорости излучательного перехода S0 ← T акцепторов энергии.
В случае, когда бимолекулярными процессами можно пренебречь, распад триплетных состояний, а следовательно, и затухание фосфоресценции (в отсутствие реабсорбции излучения), происходит по экспоненциальному закону. При выполнении этих условий для обычной фосфоресценции, влияние донора на константы скоростей излучательной и безызлучательной дезактивации триплетных молекул акцептора должно проявляться либо в изменении времени затухания сенсибилизированной фосфоресценции при сохранении экспоненциального характера кинетики, либо в отступлении от экспоненциального закона затухания. Поэтому прежде всего необходимо было экспериментально установить закон по которому происходит затухание сенсибилизированной фосфоресценции для всех систем, используемых в работе, и сравнить время затухания сенсибилизированной фосфоресценции и обычной при одних и тех же концентрациях раствора.
Методика
определения константы скорости излучательной дезактивации триплетных молекул
акцептора ![]() при их сенсибилизированном
возбуждении основана на следующих рассуждениях.
при их сенсибилизированном
возбуждении основана на следующих рассуждениях.
Для
стационарной квантовой интенсивности (число квантов испускаемых в единицу
времени) фосфоресценции ![]() можно
записать следующее выражение
можно
записать следующее выражение
![]() , (2.1)
, (2.1)
где ![]() –
константа скорости спонтанных переходов молекулы из триплетного состояния на
все электронно-колебательные уровни основного состояния;
–
константа скорости спонтанных переходов молекулы из триплетного состояния на
все электронно-колебательные уровни основного состояния; ![]() – число триплетных молекул
в стационарном режиме.
– число триплетных молекул
в стационарном режиме.
Доля молекул в
триплетном состоянии определяется с учётом времени разгорания и затухания
фосфоресценции [17,66-69], и учитывая что число триплетных молекул в условиях
стационарного возбуждения можно представить как ![]() ,
где q- доля молекул в триплетном состоянии от общего их
числа N, выражение (2.1) перепишется в виде
,
где q- доля молекул в триплетном состоянии от общего их
числа N, выражение (2.1) перепишется в виде
![]() , (2.2)
, (2.2)
Допустим, мы
имеем одно вещество с известным значением константы скорости излучательной
дезактивации энергии триплетных состояний ![]() ,
а другое вещество, то для которого эту величину необходимо определить. Тогда
для отношения их квантовых интенсивностей фосфоресценции согласно (2.2) можно
записать
,
а другое вещество, то для которого эту величину необходимо определить. Тогда
для отношения их квантовых интенсивностей фосфоресценции согласно (2.2) можно
записать
 . (2.3)
. (2.3)
Величины с индексом 0 относятся к молекулам, для которых константа скорости излучательной дезактивации энергии триплетных возбуждений известна, а величины без индекса относятся к молекулам, для которых знание константы скорости излучательного перехода необходимо определить.
Если взять одинаковые объемы раствора с равными концентрациями обоих веществ и создать условия, при которых все молекулы участвуют в излучении, то (2.3) преобразуется в
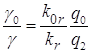 . (2.4)
. (2.4)
Величины ![]() и
и ![]() можно определить
экспериментально из кинетики разгорания и затухания фосфоресценции по формуле
можно определить
экспериментально из кинетики разгорания и затухания фосфоресценции по формуле 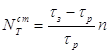 , предложенной Алфимовым с
сотрудниками [70,71].
, предложенной Алфимовым с
сотрудниками [70,71].
Величину ![]() можно определить
графически. Эта величина равна отношению площадей под спектрами фосфоресценции
исследуемых соединений
можно определить
графически. Эта величина равна отношению площадей под спектрами фосфоресценции
исследуемых соединений ![]() , записанных при
одних и тех же параметрах экспериментальной установки (спектральная ширина
щелей, напряжение на ФЭУ, усиление и т.д.).
, записанных при
одних и тех же параметрах экспериментальной установки (спектральная ширина
щелей, напряжение на ФЭУ, усиление и т.д.).
Следует отметить, что если даже неизвестны константы скоростей для обоих веществ, то таким образом, с использованием формулы (2.4), можно установить как и во сколько раз они отличаются.
Таким же образом можно определить и константу скорости излучательной дезактивации триплетных молекул акцептора при их сенсибилизированном возбуждении, если она известна для данного соединения при обычном возбуждении, в отсутствие донора. Тогда в формуле (2.4) величины с индексом 0 будут относиться к обычному возбуждению, а без индекса к сенсибилизированному. Следует подчеркнуть, что число молекул акцептора, участвующих в обычной и сенсибилизированной фосфоресценции будет одинаковым при выполнении двух необходимых условий. Первое условие – это необходимость равенства концентраций молекул акцептора в растворе в обоих случаях. Второе условие – это то, чтобы все молекулы акцептора находились в сфере тушения донора, а значит и участвовали в излучении СФ. Выполнения последнего условия можно добиться взяв эквимолярные растворы донорно-акцепторной смеси с суммарной концентрацией компонент, обеспечивающей среднее расстояние между молекулами донора и акцептора не более 1,5 нм.
Ниже изложенная методика была применена для определения констант скоростей излучательной дезактивации триплетных молекул нафталина и аценафтена в присутствии донора энергии – бензофенона.
Глава III. Влияние донора на константу скорости излучательного перехода в молекулах акцептора.
3.1 Зависимость константы скорости излучательного перехода триплетных молекул акцептора от концентрации донорно-акцепторной смеси.
Для определения константы скорости излучательного перехода молекул нафталина в присутствии бензофенона использовались эквимолярные растворы донорно-акцепторной смеси с концентрациями компонент от 0,2 до 0,5 моль/л. При таких концентрациях эквимолярного раствора можно считать, что все молекулы нафталина находятся в радиусе обменных взаимодействий с бензофеноном и, следовательно, участвуют в излучении сенсибилизированной фосфоресценции. С учетом термического сжатия растворителя, при его охлаждении от комнатной температуры до температуры кипения жидкого азота (77 К), среднее расстояние между молекулами компонент донорно-акцепторной пары, при изменении концентрации раствора в указанных выше пределах, изменялось от 14,0 до 10,3Å.
Изменение объема раствора в результате его охлаждения в указанном выше интервале температур определялось экспериментально. При этом отношение объема раствора при комнатной температуре к его объему при 77К равнялось в среднем 1,4.
Для нафталина,
константа скорости излучательной дезактивации триплетных молекул известна [29]
и равна ![]() . У бензофенона константа
скорости излучательной дезактивации триплетных молекул [29] равна
. У бензофенона константа
скорости излучательной дезактивации триплетных молекул [29] равна ![]() .
.
Значения
константы скорости излучательной дезактивации триплетных молекул нафталина ![]() при их сенсибилизированном
возбуждении, определенные экспериментально, приведены в таблице 3.1. Там же
приведены значения этой величины рассчитанной по формуле
при их сенсибилизированном
возбуждении, определенные экспериментально, приведены в таблице 3.1. Там же
приведены значения этой величины рассчитанной по формуле
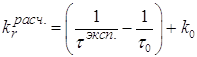 , (3.1)
, (3.1)
где ![]() и
и
![]() – времена затухания
обычной и СФ при одной и той же концентрации нафталина в растворе.
– времена затухания
обычной и СФ при одной и той же концентрации нафталина в растворе.
Таблица 3.1
Константа скорости излучательного перехода триплетных молекул нафталина в присутствии бензофенона в толуоле при 77К.
|
Концентрация |
Расстояние в донорно-акцепторной паре R, Å | Константа скорости излучательного перехода | |
|
|
|
||
|
0,2 0,3 0,4 0,5 |
14,0 12,3 11,1 10,3 |
0,024 0,031 0,039 0,047 |
0,025 0,028 0,121 0,515 |
В процессе экспериментальных исследований было обнаружено увеличение константы скорости излучательной дезактивации триплетных молекул нафталина при их сенсибилизированном возбуждении, обусловленное взаимодействием между триплетными молекулами нафталина и молекулами бензофенона в основном состоянии. Как видно из таблицы 3.1, это увеличение тем больше, чем меньше расстояние между молекулами в донорно-акцепторной паре (больше концентрация раствора).
Выражение (3.1)
записано в предположении, что уменьшение времени затухания фосфоресценции
нафталина происходит только за счет роста константы скорости излучательной
дезактивации триплетных молекул нафталина. Если же уменьшение времени затухания
СФ происходит еще в результате увеличения вероятности безызлучательной
дезактивации триплетных молекул нафталина, то значение величины ![]() , вычисленное по формуле
(3.1), должно быть больше чем
, вычисленное по формуле
(3.1), должно быть больше чем ![]() . Как
видно из таблицы 3.1 для концентраций раствора 0,3 моль/л и меньших значения
величин
. Как
видно из таблицы 3.1 для концентраций раствора 0,3 моль/л и меньших значения
величин ![]() и
и ![]() различаются незначительно.
Это различие не превышает ошибки эксперимента. Следовательно, можно считать,
что уменьшение времени затухания фосфоресценции нафталина при добавлении
бензофенона в раствор происходит в этом случае в основном за счет увеличения
константы скорости излучательной дезактивации их триплетных состояний. Среднее
расстояние между молекулами нафталина в растворе при этом не превышает 15,4
Å.
различаются незначительно.
Это различие не превышает ошибки эксперимента. Следовательно, можно считать,
что уменьшение времени затухания фосфоресценции нафталина при добавлении
бензофенона в раствор происходит в этом случае в основном за счет увеличения
константы скорости излучательной дезактивации их триплетных состояний. Среднее
расстояние между молекулами нафталина в растворе при этом не превышает 15,4
Å.
Для
концентраций компонент в эквимолярных растворах больших, чем 0,3 моль/л
величина ![]() начинает значительно
превышать
начинает значительно
превышать ![]() . Это значит, что при
средних расстояниях между молекулами нафталина в донорно-акцепторной смеси
меньших, чем 14 Å, существенный вклад в уменьшение времени затухания
сенсибилизированной фосфоресценции вносит увеличение константы скорости
безызлучательной дезактивации триплетных молекул нафталина.
. Это значит, что при
средних расстояниях между молекулами нафталина в донорно-акцепторной смеси
меньших, чем 14 Å, существенный вклад в уменьшение времени затухания
сенсибилизированной фосфоресценции вносит увеличение константы скорости
безызлучательной дезактивации триплетных молекул нафталина.
Была предпринята попытка установить характер зависимости константы скорости излучательного перехода в молекулах нафталина от среднего расстояния между компонентами донорно-акцепторной смеси. Если предположить, что увеличение вероятности излучательного перехода в молекулах акцептора при их сенсибилизированном возбуждении обусловлено обменными взаимодействиями между молекулой донора в основном состоянии и триплетной молекулой акцептора, то можно ожидать, что величина этого изменения будет пропорциональна величине обменных взаимодействий. Величина же обменных взаимодействий пропорциональна плотности перекрываемых электронных облаков, которая экспоненциально убывает на периферии. Это давало основания искать данную зависимость в экспоненциальном виде.
На рис. 3.1
представлена зависимость константы скорости излучательной дезактивации
триплетных молекул от среднего расстояния между компонентами
донорно-акцепторной пары в координатах ![]() .
.

Рис. 3.1 Зависимость константы скорости излучательной дезактивации триплетных молекул нафталина от расстояния в паре
Как видно из рисунка 3.1, экспериментальные точки хорошо укладываются на экспоненту (сплошная линия) вида
![]() . (3.2)
. (3.2)
Здесь ![]() – константа скорости
излучательной дезактивации триплетных молекул нафталина, в отсутствие
бензофенона в растворе;
– константа скорости
излучательной дезактивации триплетных молекул нафталина, в отсутствие
бензофенона в растворе; ![]() и
и ![]() – некоторые константы. При
– некоторые константы. При
![]()
![]() , что в пределах ошибки
измерений совпадает с величиной
, что в пределах ошибки
измерений совпадает с величиной![]() . Здесь
. Здесь ![]() константа скорости
излучательной дезактивации триплетных молекул бензофенона в отсутствие
нафталина в растворе. Поэтому для
константа скорости
излучательной дезактивации триплетных молекул бензофенона в отсутствие
нафталина в растворе. Поэтому для ![]() в
данном случае можно записать
в
данном случае можно записать
![]() (3.3)
(3.3)
С учетом (3.3) выражение (3.2) можно переписать в виде
![]() (3.4)
(3.4)
Таким образом,
результаты исследования зависимости константы скорости излучательной
дезактивации молекул нафталина ![]() от
среднего расстояния между компонентами в донорно-акцепторной паре
от
среднего расстояния между компонентами в донорно-акцепторной паре ![]() , когда донором является
бензофенон, показали, что
, когда донором является
бензофенон, показали, что ![]() экспоненциально
возрастает с уменьшением
экспоненциально
возрастает с уменьшением ![]() .
Характер этой зависимости удовлетворительно описывается уравнением (3.4).
.
Характер этой зависимости удовлетворительно описывается уравнением (3.4).
С целью проверки общности установленного влияния молекул бензофенона на вероятность излучательной дезактивации триплетных молекул нафталина в работе были проведены подобные исследования для другой донорно-акцепторной пары бензофенон-аценафтен.
Люминесцентные
характеристики аценафтена близки к подобным характеристикам нафталина (см.
2.4). В литературе не удалось обнаружить информацию о значении константы
скорости излучательной дезактивации триплетных молекул аценафтена, поэтому ее
величина была определена экспериментально. В качестве вещества с известным
значением ![]() использовался нафталин.
Экспериментальные данные полученные в этом опыте представлены в таблице 3.2.
использовался нафталин.
Экспериментальные данные полученные в этом опыте представлены в таблице 3.2.
Таблица 3.2
Результаты определения константы скорости излучательной дезактивации триплетных молекул аценафтена, в отсутствие донора энергии (индексы 0 – относятся к нафталину)
| Концентрация С, моль/л | Заселенность T-уровня отн. ед. |
Относительная
интенсивность |
Константа |
|
| Аценафтен | Нафталин | |||
| 0,25 | 0,28 | 0,15 | 0,20 | 0,0017 |
| 0,50 | 0,24 | 0,13 | 0,22 | 0,0019 |
Как видно из
таблицы 3.2 среднее значение константы скорости излучательной дезактивации
триплетных молекул аценафтена ![]() .
.
При
сенсибилизированном возбуждении экспериментально определялось отношение
константы скорости излучательной дезактивации триплетных молекул аценафтена ![]() эквимолярного раствора при
их сенсибилизированном возбуждении к значению этой величины
эквимолярного раствора при
их сенсибилизированном возбуждении к значению этой величины ![]() в отсутствие донора (
в отсутствие донора (![]() ).
).
Результаты этих измерений приведены в таблице 3.3. Там же приведены значения среднего расстояния между молекулами в донорно-акцепторной паре для соответствующих концентраций раствора.
Таблица 3.3
Относительное
увеличение ![]() константы скорости
излучательной дезактивации триплетных молекул аценафтена при
сенсибилизированном возбуждении для различных концентраций раствора (
константы скорости
излучательной дезактивации триплетных молекул аценафтена при
сенсибилизированном возбуждении для различных концентраций раствора (![]() ).
).
| Концентрация компонент в растворе С, моль/л | 0,2 | 0,25 | 0,3 | 0,4 | 0,5 |
| Среднее расстояние между молекулами компонентов смеси Å | 14,0 | 13,0 | 12,3 | 11,1 | 10,3 |
|
Относительное изменение
константы |
2,6 | 3,4 | 4,3 | 6,2 | 8,0 |
Как видно из
таблицы 3.3, присутствие бензофенона в сфере обменных взаимодействий аценафтена
увеличивает константу скорости излучательной дезактивации триплетных молекул
последнего в несколько раз. Как и в случае с нафталином, это увеличение тем
больше, чем меньше среднее расстояние между молекулами компонент
донорно-акцепторной смеси. Однако при одинаковом среднем расстоянии между
молекулами для пары бензофенон-нафталин и бензофенон-аценафтен в последнем
случае константа скорости излучательного перехода ![]() увеличивается
в большее число раз.
увеличивается
в большее число раз.
Чтобы
установить характер изменения ![]() от
межмолекулярного расстояния в донорно-акцепторной паре бензофенон-аценафтен был
построен график зависимости относительного прироста константы скорости
от
межмолекулярного расстояния в донорно-акцепторной паре бензофенон-аценафтен был
построен график зависимости относительного прироста константы скорости ![]() от расстояния между
молекулами компонентов в растворе (рисунок 3.2).
от расстояния между
молекулами компонентов в растворе (рисунок 3.2).
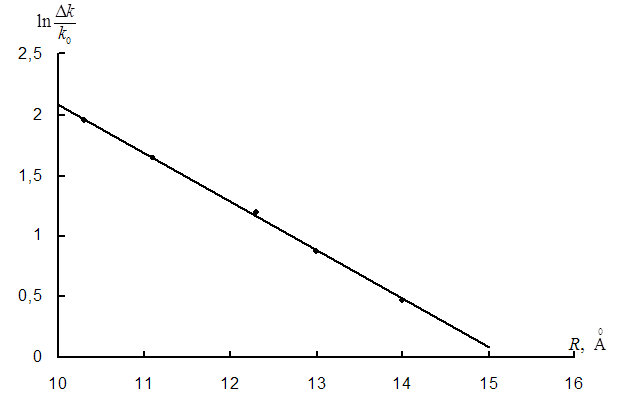
Рис. 3.2. Зависимость относительного прироста константы скорости излучательной дезактивации триплетных молекул аценафтена от среднего расстояния между молекулами компонент донорно-акцепторной смеси в толуоле при 77К.
На графике по оси абсцисс отложено среднее расстояние  между молекулами
бензофенона и аценафтена в стеклообразном толуоле при 77 К, а по оси ординат
натуральный логарифм отношения прироста
между молекулами
бензофенона и аценафтена в стеклообразном толуоле при 77 К, а по оси ординат
натуральный логарифм отношения прироста  значения
константы скорости излучательной дезактивации триплетных молекул аценафтена
значения
константы скорости излучательной дезактивации триплетных молекул аценафтена 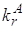 в присутствии молекул
бензофенона к ее значению
в присутствии молекул
бензофенона к ее значению 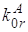 в
отсутствие молекул донора в растворе. Такой выбор системы координат обусловлен
предположением, что ожидаемая зависимость константы скорости излучательной
дезактивации триплетных молекул акцептора
в
отсутствие молекул донора в растворе. Такой выбор системы координат обусловлен
предположением, что ожидаемая зависимость константы скорости излучательной
дезактивации триплетных молекул акцептора 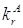 от
среднего расстояния R
описывается уравнением (3.2). Действительно, как видно из рисунка 3.2,
экспериментальные точки хорошо укладываются на экспоненту (сплошная линия),
уравнение которой имеет вид (3.2).
от
среднего расстояния R
описывается уравнением (3.2). Действительно, как видно из рисунка 3.2,
экспериментальные точки хорошо укладываются на экспоненту (сплошная линия),
уравнение которой имеет вид (3.2).
Поэтому для аценафтена можно переписать (3.2)
![]() . (3.2а)
. (3.2а)
Как и для нафталина константа ![]() для аценафтена равна
максимальному изменению величины
для аценафтена равна
максимальному изменению величины ![]() при
при ![]() . Графически, путем
экстраполяции графика представленного на рисунке 3.2 для аценафтена, когда
донором является бензофенон, было получено ее значение равное
. Графически, путем
экстраполяции графика представленного на рисунке 3.2 для аценафтена, когда
донором является бензофенон, было получено ее значение равное ![]() . Эта величина в пределах
ошибки измерения так же совпадает с величиной
. Эта величина в пределах
ошибки измерения так же совпадает с величиной ![]() .
Здесь, как и в случае с нафталином,
.
Здесь, как и в случае с нафталином, ![]() константа
скорости излучательной дезактивации триплетных молекул бензофенона, в
отсутствие молекул акцептора в растворе. Таким образом и в данном случае
величина
константа
скорости излучательной дезактивации триплетных молекул бензофенона, в
отсутствие молекул акцептора в растворе. Таким образом и в данном случае
величина ![]() определяется выражением
определяется выражением
![]() . (3.3а)
. (3.3а)
На основании этих экспериментальных результатов можно выражение (3.4) переписать для аценафтена
![]() . (3.4а)
. (3.4а)
Величина ![]() в (3.4) и (3.4а)
характеризует быстроту увеличения
в (3.4) и (3.4а)
характеризует быстроту увеличения ![]() с
уменьшением среднего межмолекулярного расстояния между компонентами
донорно-акцепторной смеси. Ее значения, определенные из графиков рис. 3.1 и
рис. 3.2 для нафталина и аценафтена соответственно равны
с
уменьшением среднего межмолекулярного расстояния между компонентами
донорно-акцепторной смеси. Ее значения, определенные из графиков рис. 3.1 и
рис. 3.2 для нафталина и аценафтена соответственно равны ![]() =3,80 нм-1 и
=3,80 нм-1 и ![]() =3,65 нм-1.
=3,65 нм-1.
Если известна
величина ![]() , то значение
, то значение ![]() для молекул акцептора, в
отсутствие донора можно вычислить, определив экспериментально отношение
для молекул акцептора, в
отсутствие донора можно вычислить, определив экспериментально отношение ![]() и зная константу скорости
излучательного перехода для молекул донора
и зная константу скорости
излучательного перехода для молекул донора ![]() .
Действительно, разделив обе части уравнения (3.4) на
.
Действительно, разделив обе части уравнения (3.4) на ![]() имеем
имеем
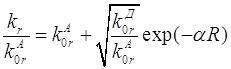 . (3.5)
. (3.5)
Здесь ![]() –
константа скорости излучательного перехода в акцепторе, а
–
константа скорости излучательного перехода в акцепторе, а ![]() – в доноре.
– в доноре.
Из (3.5) получаем
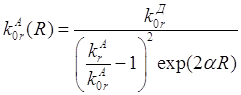 . (3.6)
. (3.6)
Как видно из (3.5), при одном и том же среднем расстоянии между компонентами донорно-акцепторной смеси, относительное изменение константы скорости излучательной дезактивации триплетных молекул акцептора тем больше, чем меньше ее абсолютное значение в отсутствие донора в растворе.
Ниже в таблице приведены значения констант скоростей излучательной дезактивации триплетных молекул нафталина и аценафтена, в отсутствие донора, рассчитанные по формуле (3.6).
Как видно из
таблицы 3.4 значение ![]() для нафталина с
точностью до
для нафталина с
точностью до ![]() совпадает с ее литературным
значением равным
совпадает с ее литературным
значением равным ![]() . Для аценафтена
разброс значений
. Для аценафтена
разброс значений ![]() рассчитанных по
формуле (3.6) немного больше, чем для нафталина и отличается от значения
определенного по методике описанной выше, с использованием формулы (2.4) не
более чем на 20%.
рассчитанных по
формуле (3.6) немного больше, чем для нафталина и отличается от значения
определенного по методике описанной выше, с использованием формулы (2.4) не
более чем на 20%.
Таблица 3.4
Значение
константы ![]() для нафталина и аценафтена
рассчитанные по формуле (3.6).
для нафталина и аценафтена
рассчитанные по формуле (3.6).
| R, Å | 10,3 | 11,1 | 12,3 | 14,0 |
| Нафталин | ||||
|
|
0,017 | 0,017 | 0,016 | 0,015 |
| Аценафтен | ||||
|
|
0,009 | 0,0018 | 0,0019 | 0,0023 |
Таким образом,
результаты исследования влияния взаимодействия между триплетными молекулами
акцептора и молекулами донора в основном состоянии на вероятность излучательной
дезактивации энергии триплетного возбуждения в акцепторе показали следующее.
Такое взаимодействие увеличивает вероятность дезактивации триплетных молекул
акцептора в системах для которых![]() . При
этом константа скорости излучательного перехода экспоненциально увеличивается с
уменьшением среднего расстояния между компонентами донорно-акцепторной смеси.
. При
этом константа скорости излучательного перехода экспоненциально увеличивается с
уменьшением среднего расстояния между компонентами донорно-акцепторной смеси.
3.2 Изменение времени затухания сенсибилизированной фосфоресценции за счёт константы скорости излучательного перехода в акцепторе.
Было показано [72-74], что затухание сенсибилизированной фосфоресценции акцептора происходит быстрее, чем при обычном его фотовозбуждении в отсутствии донора. Необходимо было выяснить, в каком случае это различие можно объяснить обнаруженным увеличением вероятности излучательного перехода в молекулах акцептора в присутствии донора. Для этого была исследована зависимость времени затухания сенсибилизированной фосфоресценции от расстояния между компонентами донорно-акцепторной смеси для эквимолярных растворов и для растворов, в которых концентрация молекул акцептора была намного меньше концентрации донора и произведено сравнение этих результатов со временем затухания, вычисленным в предположении, что его изменение обусловлено только увеличением вероятности излучательной дезактивации триплетных молекул.
Известно, что в отсутствие реабсорбции излучения, между временем затухания фосфоресценции и константами скоростей излучательной и безызлучательной дезактивации энергии триплетного возбуждения [44] существует следующая связь
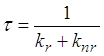 . (3.7)
. (3.7)
Здесь, как и
выше, ![]() – константа скорости
излучательной дезактивации, а
– константа скорости
излучательной дезактивации, а ![]() –
константа скорости безызлучательной дезактивации триплетных молекул. В
предположении постоянства величины
–
константа скорости безызлучательной дезактивации триплетных молекул. В
предположении постоянства величины ![]() , для
сенсибилизированной фосфоресценции (3.7) можно переписать в виде
, для
сенсибилизированной фосфоресценции (3.7) можно переписать в виде
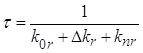 , (3.8)
, (3.8)
где ![]() –
константа скорости излучательной дезактивации триплетных молекул акцептора в
отсутствие донора;
–
константа скорости излучательной дезактивации триплетных молекул акцептора в
отсутствие донора; ![]() – изменение
константы скорости излучательного перехода в акцепторе за счет взаимодействия
между триплетной молекулой акцептора и молекулами донора в основном состоянии.
В отсутствие донора в растворе
– изменение
константы скорости излучательного перехода в акцепторе за счет взаимодействия
между триплетной молекулой акцептора и молекулами донора в основном состоянии.
В отсутствие донора в растворе ![]() и время
затухания обычной фосфоресценции равно
и время
затухания обычной фосфоресценции равно
![]() . (3.9)
. (3.9)
С учетом (3.9) выражение (3.8) можно переписать в виде
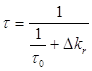 , (3.10)
, (3.10)
или, обозначив ![]() , окончательно имеем
, окончательно имеем
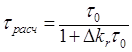 . (3.11)
. (3.11)
С учетом изменения вероятности безызлучательного перехода (3.10) можно записать в виде
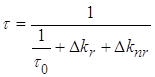 (3.10а)
(3.10а)
Здесь ![]() – изменение константы
безызлучательного перехода в молекулах акцептора.
– изменение константы
безызлучательного перехода в молекулах акцептора.
Тогда (3.11) будет иметь вид
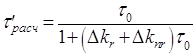 (3.11а)
(3.11а)
Если изменение
времени затухания происходит только за счет изменения вероятности
излучательного перехода, то ![]() и
и ![]() . Когда скорость затухания
фосфоресценции увеличивается как в результате роста вероятности излучательного
перехода, так и за счет увеличения вероятности безызлучательной дезактивации
триплетных молекул акцептора, тогда
. Когда скорость затухания
фосфоресценции увеличивается как в результате роста вероятности излучательного
перехода, так и за счет увеличения вероятности безызлучательной дезактивации
триплетных молекул акцептора, тогда ![]() .
.
Таким образом,
определив экспериментально время затухания сенсибилизированной фосфоресценции ![]() и обычной в отсутствие донора
и обычной в отсутствие донора
![]() фосфоресценции, и зная
фосфоресценции, и зная ![]() , можно рассчитать время
затухания сенсибилизированной фосфоресценции
, можно рассчитать время
затухания сенсибилизированной фосфоресценции ![]() в
предположении, что его отличие от
в
предположении, что его отличие от ![]() обусловлено
изменением только вероятности излучательного перехода. Экспериментально
определенное время затухания сенсибилизированной фосфоресценции
обусловлено
изменением только вероятности излучательного перехода. Экспериментально
определенное время затухания сенсибилизированной фосфоресценции ![]() включает в себя изменение
как излучательной, так и безызлучательной вероятности перехода и поэтому, в
общем случае, равно
включает в себя изменение
как излучательной, так и безызлучательной вероятности перехода и поэтому, в
общем случае, равно ![]() . Поэтому, если
при добавлении донора в раствор время затухания сенсибилизированной
фосфоресценции уменьшается только за счет роста вероятности излучательного
перехода
. Поэтому, если
при добавлении донора в раствор время затухания сенсибилизированной
фосфоресценции уменьшается только за счет роста вероятности излучательного
перехода ![]() , то
, то ![]() . Если же существенный
вклад в уменьшение времени затухания сенсибилизированной фосфоресценции вносит
рост константы безызлучательной дезактивации триплетных молекул акцептора в
результате появления дополнительных каналов деградации энергии, то
. Если же существенный
вклад в уменьшение времени затухания сенсибилизированной фосфоресценции вносит
рост константы безызлучательной дезактивации триплетных молекул акцептора в
результате появления дополнительных каналов деградации энергии, то ![]() .
.
В таблице 3.5 приведены результаты таких измерений для пары бензофенон-нафталин в толуоле при 77 K. Измерения производились для эквимолярных растворов в интервале концентраций компонент от 0,2 до 0,5 моль/л. Такой выбор относительной концентрации компонент позволил сделать вывод о роли миграции энергии по молекулам нафталина в изменении ее времени затухания.
Таблица 3.5
Время затухания сенсибилизированной фосфоресценции нафталина, определенное экспериментально и рассчитанное по формуле (3.11).
Концентрация компонент в растворе, моль/л |
Расстояние между молекулами акцептора, Å | Время затухания фосфоресценции, с | |||
|
СН |
СБ |
R |
τэксп |
τрасч |
τ0 |
| 0,2 | 0,2 | 17,7 | 2,30 | 2,30 | 2,35 |
| 0,3 | 0,3 | 15,4 | 2,24 | 2,23 | 2,30 |
| 0,4 | 0,4 | 14,0 | 1,70 | 1,98 | 2,07 |
| 0,5 | 0,5 | 13,0 | 0,82 | 1,39 | 1,45 |
| 0,05 | 0,5 | 28,2 | 2,28 | 2,29 | 2,35 |
Как видно из
таблицы 3.5, если среднее расстояние между молекулами нафталина больше 1,5 нм,
то ![]() . Это значит, что в этом
случае уменьшение времени затухания сенсибилизированной фосфоресценции
нафталина в сравнении с обычной обусловлено увеличением константы скорости
излучательной дезактивации триплетных молекул. Когда среднее расстояние между
молекулами нафталина больше 1,5 нм, то
. Это значит, что в этом
случае уменьшение времени затухания сенсибилизированной фосфоресценции
нафталина в сравнении с обычной обусловлено увеличением константы скорости
излучательной дезактивации триплетных молекул. Когда среднее расстояние между
молекулами нафталина больше 1,5 нм, то ![]() .
Это значит, что при концентрациях молекул нафталина в растворе для которых R<1,5 нм, появляются дополнительные каналы
безызлучательной дезактивации его триплетных возбуждений. При таких значениях R создаются благоприятные условия для миграции триплетных
возбуждений по молекулам нафталина и становится актуальным
миграционно-ускоренное тушение на различного рода тушителях.
.
Это значит, что при концентрациях молекул нафталина в растворе для которых R<1,5 нм, появляются дополнительные каналы
безызлучательной дезактивации его триплетных возбуждений. При таких значениях R создаются благоприятные условия для миграции триплетных
возбуждений по молекулам нафталина и становится актуальным
миграционно-ускоренное тушение на различного рода тушителях.
Таким образом, для таких соединений как нафталин, у которых вероятность излучательной дезактивации энергии триплетных возбуждений намного меньше вероятности ее безызлучательной дезактивации, вклад роста константы скорости излучательного перехода в изменение времени затухания фосфоресценции акцептора при добавлении донора в раствор невелик. Однако, при концентрациях акцептора в растворе 0,3 моль/л и меньших, различие в кинетике сенсибилизированной фосфоресценции обусловлено именно этим механизмом. Это различие хотя и невелико, но превышает ошибку эксперимента.
Для эквимолярных растворов с концентрацией компонент больших 0,3моль/л, существенный вклад в изменение времени затухания фосфоресценции нафталина вносит миграционно-ускоренное тушение его триплетных состояний. Так для концентраций компонент в растворе 0,5моль/л время затухания сенсибилизированной фосфоресценции нафталина уменьшается в сравнении с временем затухания обычной фосфоресценции только в 1,04 раза за счет роста вероятности излучательного перехода в 2,9 раза и уменьшается в 1,7 раза за счет появления дополнительных каналов безызлучательной дезактивации.
Нами были проведены измерения τэксп, τрасч, τ0 так же и для аценафтена в стеклообразном толуоле при 77 K. Концентрация аценафтена в растворе как при сенсибилизированном, так и при обычном возбуждении равнялась 0,5 моль/л. Концентрация бензофенона при сенсибилизированном возбуждении составляла 0,5 моль/л. Результаты этих измерений приведены в таблице 3.6.
Таблица 3.6
Время затухания обычной τ0 и сенсибилизированной фосфоресценции нафталина, определенное экспериментально τэксп и рассчитанное τрасч по формуле (3.11)
| Расстояние между молекулами аценафтена R, Å | Время затухания фосфоресценции, с | ||
|
τэксп |
τрасч |
τ0 |
|
| 13,0 | 1,55 | 2,05 | 2,10 |
Как видно из таблицы 3.6 время затухания сенсибилизированной фосфоресценции аценафтена уменьшается в 1,024 раза за счет увеличения вероятности излучательного перехода в этом случае. Его изменение в результате увеличения вероятности безызлучательной дезактивации триплетных молекул при этом происходит в 1,32 раза. Как видно, при данной концентрации эквимолярного раствора донорно-акцепторной смеси бензофенон-аценафтен, вклад обоих указанных выше механизмов в уменьшение времени затухания сенсибилизированной фосфоресценции в сравнении с обычной различен. Уменьшение времени затухания сенсибилизированной фосфоресценции аценафтена происходит в основном за счет появления дополнительных каналов безызлучательной дезактивации триплетных возбуждений.
Основные результаты и выводы.
В результате проделанной работы были получены следующие основные результаты:
1. Разработана методика определения константы скорости излучательного перехода молекул из триплетного состояния в основное. Эта методика сравнительно проста и основана на измерении интенсивности и заселенности триплетного уровня вещества, для которого определяется данная величина по отношению к интенсивности фосфоресценции и заселенности триплетного уровня вещества с известным значением константы скорости излучательного перехода.
Данная методика может быть использована в случае, когда заселенность триплетного уровня высока. Поэтому она применима только для твердых растворов органических соединений.
2. Показано, что присутствие донора в радиусе обменных взаимодействий с триплетными молекулами акцептора, увеличивает константу скорости излучательной дезактивации последних. Относительное изменение вероятности излучательного перехода в молекулах акцептора экспоненциально зависит от среднего расстояния между компонентами донорно-акцепторной смеси и от соотношения соответствующих констант скоростей переходов молекул акцептора и донора энергии.
3. Установлено, что уменьшение времени затухания сенсибилизированной фосфоресценции в сравнении с обычной, в отсутствие молекул донора, обусловлено увеличением констант скоростей как излучательной, так и безызлучательной дезактивации триплетных молекул акцепторов при добавлении донора в раствор. Относительный вклад этих двух механизмов в относительное изменение времени затухания сенсибилизированной фосфоресценции различен и зависит как от концентрации каждой из компонент в растворе, так и от их природы.
Эти результаты позволили сделать следующие выводы:
1. Наблюдаемая
более высокая интенсивность сенсибилизированной фосфоресценции в сравнении с
обычной таких соединений как нафталин, аценафтен в твердых растворах при 77 K обусловлена не более эффективным заселением их триплетных
состояний при сенсибилизированном возбуждении, а увеличением константы скорости
излучательной дезактивации их триплетных состояний в присутствии донора.
Следует подчеркнуть, что этот вывод относится к случаю, который часто
реализуется на практике, если константа скорости излучательного перехода для
молекул донора намного больше соответствующей константы для молекул акцептора (![]() ).
).
2. Взаимодействия между молекулами акцепторов в триплетном состоянии и молекулами доноров в основном состоянии возмущают соответствующие электронные состояния, что приводит к увеличению излучательной константы скорости электронного перехода в акцепторе. Этот результат указывает на ошибочность основного положения теории Ферстера-Декстера переноса энергии [1,11] по обменно-резонансному механизму, где для приготовления начального и конечного квантовых состояний донорно-акцепторной пары берутся невозмущенные волновые функции донора и акцептора в соответствующих состояниях, и находится в качественном соответствии с выводами новой теории переноса энергии [14,15,22,23].
3. Более быстрая дезактивация триплетных молекул акцептора в присутствии донора обусловлена ростом как излучательной, так и безызлучательной константы скоростей дезактивации возбуждений. Влияние концентрации на изменение вероятности излучательного перехода определяется только изменением среднего расстояния между молекулами различных компонент донорно-акцепторной смеси.
СПИСОК ЛИТЕРАТУРЫ
1. Ермолаев В.Л., Бодунов Е.Н., Свешникова Е.Н., Шахвердов Т.И. Безызлучательный перенос энергии электронного возбуждения. – Л.: Наука, 1977 – 311с.
2. Агранович В.М., Галанин М.Д. Перенос энергии электронного возбуждения в конденсированных средах. – М.: Наука, 1978 – 384с.
3. Spectroscopy and Excitation Dynamics of Condensed Molecular System / Eds. Agranovich V.H., Hochstraser R.M. – Amsterdam: North – Holland, 1983. Спектроскопия и динамика возбуждений в конденсированных молекулярных системах / Под ред. Аграновича В.М. и Хохштрассера Р.М. – М, 1987 – 492с.
4. Теренин А.Н. Фотоника молекул красителей. – Л.: Наука, 1967 – 616 с.
5. Паркер С. Фотолюминесценция растворов. – М.: Мир, – 1972 – 511с.
6. Красновский А.А. Фотохимия хлорофилла и его аналогов/ В сб. элементарные фотопроцессы в молекулах – М.: Наука. – 1966. – С. 213 – 242.
7. Портер Дж. Профессор Александр Теренин (1896 – 1967) – пионер фотохимии. К 100 – летию со дня рождения// Оптика и спектроскопия. – 1997. – Т.83. – №4. – С. 534 – 538.
8. Гурвич А.М. Введение в физическую химию кристаллофосфоров – М.: Высшая школа, 1982 – 376 с.
9. Багнич С.А. Миграция триплетных возбуждений сложных молекул в неупорядоченных средах и системах с ограниченной геометрией (обзор)// Физика твердого тела. – 2000. – Т.42. – №10. – С. 1729 – 1756.
10. Förster Th. Naturforsch Z. Untersuchung des zwischenmolekularen ubergangs von Electronenanregungsenergie. – 1949. – 4a – №50 – S.321–327.
11. Dexter D.L. Theory of sensitized luminescence in solids // J. Chem. Phys. – 1953. – V.21. – №5 – P.836 – 850.
12. Артюхов В.Я., Майер Г.В. Теория переноса энергии электронного возбуждения в сложных молекулярных системах// Известия вузов. Физика. – 2000. – №10–С. 24 – 29.
13. Артюхов В.Я., Майер Г.В. Квантово – химическая теория переноса энергии электронного возбуждения в молекулярных системах// Журнал физической химии. – 2001. –Т.75. – №6. – С.1143–1150.
14. Артюхов В.Я., Майер Г.В., Риб Н.Р.. Квантово-химическое исследование триплет-триплетного переноса энергии электронного возбуждения в бихромоформных молекулярных системах // Оптика и спектроскопия. – 1997. – Т.83. – №5. – С.743 – 748.
15. Артюхов В.Я., Майер Г.В. Электронные состояния и фотопроцессы в бихромоформных системах// Журнал прикладной спектроскопии. – 2002. – Т.69. – №2. – С.172 – 180.
16. Вашкевич О.В., Голубин М.А., Дерябин М.И. О причинах смещения и уширения полос спектров фосфоресценции твёрдых растворов органических соединений// Вестник Северо – Кавказского государственного технического университета. Серия «Физико – химическая». – 2003 - №1(7) . – С. 49-51.
17. Дерябин М.И., Глушков А.В., Шальнев А.Ю. Влияние температуры на параметры фосфоресценции и поглощения донора энергии в замороженных парафиновых растворах// Известия высших учебных заведений. Физика. – 2003 - №7 . С. 6-9
18. Вашкевич О.В., Голубин М.А., Дерябин М.И. Миграционно – ускоренное тушение кислородом возбуждений примесных центров в стёклах при 77 К// III – Международная научная конференция «Химия твёрдого тела и современные микро- и нанотехнологии» Кисловодск. – 2003 – С. 21-22
19. Дерябин М.И., Куликова О.И. Влияние температуры на концентрационное тушение сенсибилизированной фосфоресценции органических молекул в н.- парафиновых растворах// Журнал прикладной спектроскопии. – 2003. – Т. 70. – №6. – С. 779-783.
20. Теренин А.Н., Ермолаев В.Л. Сенсибилизированная фосфоресценция органических молекул при низких температурах// Доклады АН СССР. Физика – 1952. – Т.LXXXV. – №3 – С.547–550.
21. Ландау Л.Д., Лифшиц Е.М. Квантовая механика. – М.: Изд-во физико-математической литературы, 1963. – 704 с.
22. Артюхов В.Я., Майер Г.В. Теоретическое исследование влияния ориентации и растворителя на перенос энергии в бихромоформных системах// Оптика и спектроскопия. – 2001. – Т.90. – №5. – С.743 – 747.
23. Артюхов В.Я., Майер Г.В. Новая теория переноса электронной энергии в молекулярных системах // Международная конференция по люминесценции, посвященная 110-летию со дня рождения академика С.И. Вавилова. Тезисы докладов. – Москва, 2001 – С. 100.
24. Inokuti M. Hirayama F. Influence of energy transfer by the exchange mecanism on donor luminescence // J. Chem. Phys. – 1965. – V.43. – №6. – P.1978 – 1989.
25. Ермолаев В.Л. Сенсибилизированная фосфоресценция органических соединений: триплет-триплетный перенос энергии// Элементарные фотопроцессы в молекулах. – Л.: Наука, 1966 – С.147–162.
26. Клаверье П. // Межмолекулярные взаимодействия / Под. ред. Пюльмана Б.М. – М.: Мир, 1981 – С.99.
27. Молекулярные взаимодействия / Под. ред. Райтмана Г., Орвигл-Томоса. – М.: Мир, 1984 – 598с.
28. Майер Г.В., Артюхов В.Я., Базыль О.К. и др. Электронно-возбужденные состояния и фотохимия органических соединений.: Новосибирск – 1997 – С.76–95, 101–152.
29. Ермолаев В.Л.. Перенос энергии в органических системах с участием триплетного состояния// Успехи физических наук. – 1963. – Т.80. – №1 С.3 – 40
30. Горяева Е.М., Шабля А.В., Ермолаев В.Л. Безызлучательная дезактивация нижнего триплетного состояния нафталина и его оксипроизводных при 77К// Оптика и спектроскопия. – 2003. – Т.95. – №2. – С.198–207.
31. Шпольский Э.В. Проблемы происхождения и структуры квазилинейчатых спектров органических соединений при низких температурах// Успехи физических наук. – 1962. – Т.77. – №2. – С.321–336.
32. Доброхотова В.К., Кульчицкий В.А., Набойкин Ю.В. Спектры замороженных растворов двух примесей при 77К// Известия АН СССР. Серия физическая. – 1963. – Т.27. – №6. – С.690–692.
33. Климова Л.А., Нерсесова Г.Н. Спектры флуоресценции и поглощения бинарных смесей ароматических углеводородов в замороженных кристаллических растворах// Журнал прикладной спектроскопии. – 1965. – Т.2. – №1. – С.45–50.
34. Глядковский В.И., Климова Л.А., Нерсесова Г.Н. Спектроскопия смесей ароматических углеводородов в замороженных кристаллических растворах// Оптика и спектроскопия. – 1967. – Т.23. – №3. – С.407 – 413.
35. Cadas J.P., Courpron C., Lochet R. Transfersts á energie entre entre éħdts triplets miltien cristallin a 77K// CR.–1962.–V.254. – №4. – P.2490 – 2492.
36. Rouset A., Lochet R., Cadas J.P.Transferts á energie entre niveux triplets de la benzophenone et du naphtaline cristallisesa 77K// J. Phys.–1963.–V.24, №2. – P.2141–2143.
37. Гобов Г.В., Коношенко В.И., Нурмухаметов Р.Н. Триплет-триплетный перенос энергии в условиях эффекта Шпольского// Оптика и спектроскопия. – 1976. – Т.40. – №2. – С.406–408.
38. Гобов Г.В., Коношенко В.И. Триплет-триплетный перенос энергии в условиях эффекта Шпольского// Журнал прикладной спектроскопии. – 1978. – Т.28. – №4. – С.663–667.
39. Гобов Г.В., Юденков В.В. Спектры сенсибилизированной фосфоресценции дифениленоксида в бинарных растворителях при 77К// Электронно-колебательные спектры некоторых ароматических соединений. – Смоленск, 1975. – С. 20–23.
40. Гобов Г.В., Конашенко В.И. Спектры сенсибилизированной фосфоресценции кристаллических растворов при 77К// Оптика и спектроскопия. – 1977. – Т.43. – №6. – С.1054–1059.
41. Гребенщиков Д.М., Блужин В.Б., Дзарагазова Т.П. и др. Т-Т перенос энергии между разными примесными центрами в матрицах Шпольского// Современные аспекты тонкоструктурной и селективной спектроскопии. – М.: 1984. – С. 27–31.
42. Расколодько В.Г., Файдыш А.Н. Спектры фосфоресценции и миграция энергии триплетного уровня в кристаллах бензофенона// Известия АН СССР. Серия физическая. – 1965. – Т.29. – №8. – С. 1309–1312.
43. Болотникова Т.Н., Наумова Т.М. К вопросу о концентрационной зависимости квазилинейчатых спектров фосфоресценции// Оптика и спектроскопия. – 1963. – Т.25. – №3. – С. 460 – 463.
44. Мак-Глин С., Адзуми Т., Киносита М. Молекулярная спектроскопия триплетного состояния. – М.: Мир, 1972 – 448с.
45. Осадько И.С. Селективная спектроскопия одиночных молекул. – М.: Физматлит, 2000 – 319с.
46. Медведов Э.С., Ошеров В.И. Теория безызлучательных переходов в многоатомных молекулах. – М.: Наука, 1977. – С.7–59.
47. Гребенщиков Д.М. Кинетика фосфоресценции некоторых ароматических соединений в кристаллических растворах// Автореферат диссертации канд. физ.-мат. наук. – Москва, – 1969. – 18 с.
48. Борисевич Н.А., Казберук Д.В., Лысак Н.А. и др. Фотофизические и фотохимические релаксационные процессы в ароматических кетонах// Известия АН СССР. – Серия физическая. – 1990. – Т.54. – №3. – С.370–376.
49. Гаевский А.С., Расколодько В.Н. и Файдыш А.Н. Влияние фазового состояния на фосфоресценцию бензофенона и передача энергии электронного возбуждения в твердых растворах// Оптика и спектроскопия. – 1967. – Т.22. – №2. – С. 232 – 239.
50. Гаевский А.С., Нелипович К.И., Файдыш А.Н. Влияние условий возбуждения и структуры решетки на миграцию и аннигиляцию триплетных экситонов в кристаллах бензофенона// Известия АН СССР. – Серия физическая. – 1973. – Т.37. – №3. – С. 423 – 500.
51. Мельник В.И., Нелипович К.И., Шпак М.Т. Особенности фосфоресценции различных модификаций бензофенона// Известия АН СССР. – Серия физическая – 1980. – Т.44, №4 – С.827–832.
52. Graham Daniel J., Labrake Dwayne L. Molecular-lever crystallization of benzophenone: Low-temperature quench, annealing and phosphorescence// J. Chem. Phys. – 1993. – V.97. – №21. – P.5594–5598.
53. Ильчимин И.П., Мельник В.И., Нелипович К.И., Шпак М.Т. Фосфоресценция X-модификации и аморфных пленок бензофенона// Журнал прикладной спектроскопии. – 1991. – Т.55. – №2. – С.811–815.
54. Гаевский А.С., Давыдова Н.А., Добровольская О.В. и др. Миграция энергии электронного возбуждения и фотореакции в жидких фазах бензофенона// Известия АН СССР. – Серия физическая. – 1970. – Т.34. – №3. – С.499–506.
55. Залесская Г.А., Яковлев Д.А., Грушевская С.А. Столкновительная передача колебательной энергии в парах и смесях бензофенона с посторонними газами// Журнал прикладной спектроскопии. – 1993. – Т.59. – №1–2. – С.32–36.
56. Королев В.В., Грицан Н.П., Хмелинский Н.В. и др. Определение параметров статического тушения фосфоресценции органических молекул по обменно-резонансному механизму// Химическая физика. – 1987. – Т.6. – №7. – С. 892 – 898.
57. Турро Н. Молекулярная фотохимия. – М.: Мир, 1967. – 328 с.
58. Горяева Е.М., Шабля А.В., Ермолаев В.Л. Роль окружения в безызлучательной дезактивации триплетных состояний производных нафталина в твердых растворах при 77 K// Оптика и спектроскопия. – 2001. – Т.90. – №90. – С. 577 – 585.
59. Левшин Л.В., Салецкий А.М. Люминесценция и ее измерения. Молекулярная люминесценция. – М.: – Изд-во МГУ, 1989. – 272с.
60. Шпольский Э.В., Климова Л.А., Нерсесова Г.Н. др. Концентрационная зависимость спектров флуоресценции и поглощения замороженных н.-парафиновых растворов нафталина// Оптика и спектроскопия. – 1968. – Т.26. – №1. – С.52–59.
61. Бурштейн А.И. Концентрационное тушение некогерентных возбуждений // Успехи физических наук. – 1984. – Т.143 – №4. – С.553 – 600.
62. Saigusa Hiroyuki, Sun Sheng, Lim E.C. Photodissociations on spectroscopy of excimers in naphthalene clusters// Phys. Cytv. – 1992. – V.96. – №25. – P. 100999 – 101001.
63. Logunov Stephan L., Rodgers Michael A.J., Subnaseonal dynamics of naphthalene – oxygen exciplex// J. Phys. Chem. – 1992. – V.96. – №7. – P.2915–2917.
64. Matsuzawa Sadao, Latotte Michel, Garrigues Phillippe and oth. Naphthalene – amines explex formation promoted by phase transition in crystallized cyclohexane// J. Phys. Chem. – 1994. – V.98. – №32. – P. 7832–7836.
65. Вентцель Е.С. Теория вероятностей. – М.: Изд-во физико-математической литературы, 1958 – 464с.
66. Дерябин М.И., Гребенщиков Д.М. Определение концентрации органических молекул в триплетном состоянии при возбуждении периодически повторяющимися импульсами. – Ред. Журнала прикладной спектроскопии. Деп. в ВННИТИ, 1988. - №7477 – В88 – 9 с.
67. Голубин М.А., Дерябин М.И., Куликова О.И. Кинетика накопления и определение числа триплетных молекул акцептора энергии в замороженных растворах.// Известия ВУЗов. Северо – Кавказский регион. Естественные науки. – 1998. - №1. – С. 52-55.
68. Дерябин М.И., Куликова О.И., Солодунов В.В. Зависимость интенсивности сенсибилизированной фосфоресценции нафталина в матрицах н.- гексана от времени отжига// Вестник Ставропольского государственного университета. – 1999. - №18. – С. 99-101 н.- гексана от времени отжига// Вестник Ставропольского государственного университета. – 1999. - №18. – С. 99-101
69. Вашкевич О.И., Дерябин М.И. Механизмы влияния концентрации и температуры на спектры и кинетику фосфоресценции органических молекул в твердых растворах// Оптический журнал. – 2004. – Т 71.– №9. – С. 12-15.
70. Алфимов М.В., Бубен Н.Я., Приступа А.Н. и др. Определение концентрации органических молекул в триплетном состоянии при возбуждении быстрыми электронами// Оптика и спектроскопия. – 1966. – Т.20. – №3. – С.424 – 426.
71. Смирнов
В.А., Алфимов М.В. Экспериментальное определение коэффициента характеризующего
вероятность перехода с ![]() для триплетных
состояний органических молекул// Кинетика и катализ. – 1966. – Т.7. – №4. – С.
583 – 588.
для триплетных
состояний органических молекул// Кинетика и катализ. – 1966. – Т.7. – №4. – С.
583 – 588.
72. Дерябин М.И., Тищенко А.Б. О концентрационной зависимости квантового выхода сенсибилизированной фосфоресценции нафталина в толуоле при 77 К// Известия высших учебных заведений. Физика. – 2004. – №10
73. Дерябин М.И., Каргин Н. И., Голубин М. А. Процессы дезактивации триплетных возбуждений примесных центров в твердых растворах органических соединений при сенсибилизированном возбуждении // IV Международная конференция «Химия твердого тела и современные микро- и нанотехнологии». – Кисловодск. – 2004
74. Дерябин М.И., Голубин М.А., Тищенко А.Б. Влияние концентрации на константу скорости излучательной дезактивации триплетных молекул аценафтена при сенсибилизированном возбуждении. Вестник Сев. Кав. ГТУ. – 2004. – №1(8) . – С. 38 – 41.