Дипломная работа: Влияние температуры на концентрацию триплетных молекул в твердых растворах при сенсибилизированном возбуждении
СТАВРОПОЛЬСКИЙ ГОСУДАРСТВЕННЫЙ
УНИВЕРСИТЕТ
На правах рукописи
УДК 535.373.2
КУЛИКОВА Ольга Игоревна
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА КОНЦЕНТРАЦИЮ ТРИПЛЕТНЫХ МОЛЕКУЛ В ТВЕРДЫХ РАСТВОРАХ ПРИ СЕНСИБИЛИЗИРОВАННОМ ВОЗБУЖДЕНИИ
01.04.07
физика конденсированного состояния
АВТОРЕФЕРАТ
диссертации на соискание учёной степени
кандидата физико-математических наук
Ставрополь – 2001
Работа выполнена в Ставропольском государственном университете
Научные руководители: доктор физико-математических наук, профессор Несис Е.И.
кандидат физико-математических наук, доцент Дерябин М.И.
Официальные оппоненты: доктор физико-математических наук, профессор Э. Н. Мясников
доктор физико-математических наук, профессор В. В. Чеканов
Ведущая организация: Северо-Кавказский государственный технический университет, г. Ставрополь
Защита состоится «___» _____________ 2001 года в ___ часов на заседании Диссертационного Совета Д 212.208.05 по физико-математическим наукам НИИ физики при Ростовском государственном университете по адресу: г. Ростов-на-Дону, пр. Стачки 194, НИИ физики РГУ.
С диссертацией можно ознакомиться в библиотеке РГУ
Автореферат разослан «____» _________ 2001 года
Учёный секретарь Диссертационного Совета
кандидат физико-математических наук А.Н. Павлов
С О Д Е Р Ж А Н И Е
ВВЕДЕНИЕ………………………………………………………………4
ГЛАВА 1. ТУШЕНИЕ ВОЗБУЖДЕННЫХ СОСТОЯНИЙ ПРИМЕСНЫХ МОЛЕКУЛ В ТВЕРДЫХ РАСТВОРАХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ (ОБЗОР ЛИТЕРАТУРЫ) …………………………………………………………13
1.1 Межмолекулярный перенос энергии триплетного возбуждения в твёрдых растворах…………………………………………………….13
1.2 Механизмы концентрационного тушения возбужденных состояний примесных молекул в растворах……..…….…………………….…..23
1.3 Влияние температуры на взаимодействия примесных органических молекул в конденсированных средах……………………………..…34
ГЛАВА 2. МЕТОДИКА ЭКСПЕРИМЕНТАЛЬНЫХ ИССЛЕДОВАНИЙ…………………………………………..……………48
2.1 Растворители и соединения………………………………………….48
2.2 Методика эксперимента……………..……………………………….58
2.3 Определение параметров триплетного состояния молекул акцептора из кинетики сенсибилизированной фосфоресценции………………………………….63
ГЛАВА 3. ОСОБЕННОСТИ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ ПАРАМЕТРОВ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕС-ЦЕНЦИИ ПРИМЕСНЫХ МОЛЕКУЛ В ЗАМОРОЖЕННЫХ Н.-ПАРАФИНАХ………………………………………………………………..….. .73
3.1 Температурная зависимость интенсивности сенсибилизированной фосфоресценции органических молекул……………………………………………….…..73
3.2 Влияние концентрации примесей и растворителя на параметры сенсибилизированной фосфоресценции……………………………………………………….82
3.3 Необратимый характер хода кривой температурной зависимости концентрации триплетных молекул акцептора…………………………………………………..92
3.4 Влияние скорости замораживания на параметры сенсибилизированной фосфоресценции……………………………………………………………………….97
3.5 Основные результаты главы 3……………………………………………………99
ГЛАВА 4. ВЛИЯНИЕ ОТЖИГА НА КОНЦЕНТРАЦИЮ ТРИПЛЕТНЫХ МОЛЕКУЛ АКЦЕПТОРА ЭНЕРГИИ…………………………………………..101
4.1 Результаты исследования влияния отжига на интенсивность сенсибилизированной фосфоресценции и характер её температурной зависимости………………………………………………………………………………..102
4.2 Влияние отжига на спектры и кинетику сенсибилизированной фосфоресценции…..…………………………………………………………………………...107
4.3 Влияние отжига на параметры фосфоресценции донора энергии…………………………………………………………………….………….113
4.4 Исследование закона накопления числа одиночных молекул акцептора, участвующих в излучении, в процессе отжига ……………………………………...117
4.5 Определение энергии активации процесса, приводящего к увеличению числа молекул акцептора, участвующих в излучнии……………………………….…122
4.6 Основные результаты главы 4…………………………………………..…..…..125
АНАЛИЗ РЕЗУЛЬТАТОВ…………...…..……………………………….……...127
ОСНОВНЫЕ РЕЗУЛЬТАТЫ И ВЫВОДЫ………………..………………...….137
ЛИТЕРАТУРА…...………………………………………………………………..140
ВВЕДЕНИЕ
Актуальность проблемы. Фундаментальные представления о механизмах фотопроцессов, составляющих основу фотосинтеза, фотодинамических методов лечения, молекулярной электроники и других, базируются в основном на классических результатах по фотонике синтетических органических соединений в конденсированных средах [1-4]. Перенос энергии электронного возбуждения, лежащий в основе этих процессов – проблема весьма универсальная, поскольку он является промежуточным процессом между актом возбуждения электронов и теми конечными процессами, в которых энергия возбужденных электронов используется [5].
К настоящему времени накоплен и обобщен большой теоретический и экспериментальный материал по межмолекулярному триплет-триплетному (Т-Т) переносу энергии электронного возбуждения [5-9]. Хорошими модельными системами, которые часто используются для экспериментального изучения и проверки выводов теории переноса энергии триплетного возбуждения между молекулами, являются твёрдые растворы органических соединений. А. Н. Теренин достаточно ясно сформулировал причину этого: «… в жестких растворах триплетные состояния являются источником долгоживущей фосфоресценции». Основные закономерности межмолекулярного Т-Т переноса энергии были установлены именно при исследовании тушения фосфоресценции молекул донора молекулами акцептора в этих системах. Однако даже для наиболее изученных донорно-акцепторных пар параметры переноса энергии триплетного возбуждения существенно отличаются у различных авторов [10-14].
Другой проблемой физики конденсированного состояния является концентрационное тушение возбужденных состояний [15-20]. Она возникает при исследовании столь разных систем, как кристаллы и стекла, активированные примесными ионами, жидкие растворы красителей и твёрдые парамагнитные растворы. Механизмы концентрационного тушения возбужденных состояний из-за сложности и многообразия возможных причин представляют значительные трудности для объяснения. Поэтому, несмотря на большое число работ, посвященных данной проблеме, ряд вопросов, связанных с ней, остается к настоящему времени нерешенным. В частности, остаются неизученными механизмы концентрационного тушения триплетных состояний примесных молекул в твердых растворах при их сенсибилизированном возбуждении. Тогда как перенос энергии по триплетным уровням примесных центров обусловлен обменными взаимодействиями, для осуществления которых необходимо перекрывание электронных облаков донорно-акцепторных пар [21]. Для создания таких условий необходимы сравнительно высокие концентрации молекул примесей в растворе, при которых заметный вклад в дезактивацию триплетных молекул может вносить их концентрационное тушение. Это может существенно влиять на достоверность определения параметров переноса энергии с использованием известных методик [10-14]. С другой стороны, знание механизмов концентрационного тушения может определить пути его снятия, что позволит целенаправленно повышать выход фотофизических и фотохимических процессов, происходящих с участием Т-Т переноса энергии.
Таким образом, изучение механизмов концентрационного тушения триплетных состояний в твердых растворах при их сенсибилизированном возбуждении имеет актуальное значение как для теории, так и для практики фотоники органических соединений в конденсированных средах.
Целью настоящей работы было установление основных механизмов концентрационного тушения триплетных молекул в твердых растворах при сенсибилизированном возбуждении, основанное на исследовании влияния температуры на их концентрацию.
Метод температурных измерений является одним из общепринятых методов исследования внутри- и межмолекулярных взаимодействий в конденсированных средах [22-25]. Изучение температурных зависимостей различных характеристик люминесценции (спектры поглощения и испускания, квантовый выход, длительности возбужденного состояния …) позволяют получить информацию о константах скоростей процессов, регулирующих накопление молекул в возбужденных состояниях и их дезактивацию. Что, в свою очередь, определяет концентрацию молекул в возбужденных состояниях в условиях стационарного возбуждения. Кроме того, изменение температуры по-разному сказывается на различных механизмах концентрационного тушения возбужденных состояний.
Поэтому изучение влияния температуры на параметры фосфоресценции молекул донорно-акцепторной смеси было выбрано в качестве основного метода исследований.
Предметом исследования были выбраны ароматические углеводороды в н.-парафиновых растворах.
В н.-парафинах примесные молекулы вытесненные в межблочное пространство в процессе их замораживания могут создавать большие локальные концентрации, намного превышающие их среднюю концентрацию, которую можно получить в стеклообразных растворителях [25-27]. С одной стороны это позволяет исследовать перенос энергии при меньших расстояниях между партнерами в донорно-акцепторной паре, с другой стороны по этой же причине в данных системах должны быть более ярко выражены механизмы концентрационного тушения триплетных состояний, вследствие чего они становятся более доступными для экспериментальных исследований.
Кроме того, имеются также практические причины для изучения ароматических молекул, так как они образуют основу большинства красителей, так же как и многих биологически важных веществ.
В диссертационной работе решались следующие задачи:
1. Разработка методики определения концентрации триплетных молекул акцептора энергии.
2. Исследование характера температурной зависимости фосфоресценции донора в присутствии молекул акцептора и сенсибилизированной фосфоресценции акцептора в замороженных н.-парафиновых растворах.
3. Исследование влияния концентрации донорно-акцепторной смеси на характер температурной зависимости сенсибилизированной фосфоресценции органических молекул.
4. Изучение влияния отжига раствора на концентрацию триплетных молекул акцептора энергии.
Диссертационная работа состоит из введения, четырёх глав, заключения и списка литературы.
Первая глава носит обзорный характер. В ней приведены краткие сведения об основных направлениях исследования межмолекулярного триплет-триплетного переноса энергии в твердых средах. Приведены несоответствия параметров переноса для одних и тех же донорно-акцепторных пар у различных авторов. Дан обзор работ по температурному и концентрационному тушению триплетных состояний органических молекул в твердых растворах и влиянию температуры на эффективность переноса энергии триплетного возбуждения. Обосновывается постановка задач настоящей работы.
Во второй главе рассмотрены методические вопросы. Наряду с вопросами выбора объектов исследования и техники эксперимента решаются задачи определения концентрации триплетных молекул акцептора энергии, константы перехода молекул акцептора в триплетное состояние из изучения кинетики их накопления и распада. Решение этих задач необходимо для последующих исследований влияния температуры на концентрацию молекул акцептора энергии в триплетном состоянии.
В третьей главе приводятся результаты изучения температурной зависимости (от 77 К до точки плавления растворителя) параметров обычной фосфоресценции донора в присутствии акцептора, а так же сенсибилизированной фосфоресценции акцептора. Температурная зависимость изучена для нескольких донорно-акцепторных пар в замороженных н.-парафинах от н.-гексана до н.-декана. Обнаружен немонотонный характер температурной зависимости сенсибилизированной фосфоресценции, включающий интервалы как уменьшения, так и увеличения интенсивности. Рассматривается влияние концентрации молекул донорно-акцепторной смеси и растворителя на характер температурной зависимости. На основании полученных данных делается вывод о том, что наличие аномального участка (область увеличения интенсивности) в температурной зависимости характерна для образцов, в которых наблюдается концентрационное тушение сенсибилизированной фосфоресценции. Показано, что увеличение интенсивности сенсибилизированной фосфоресценции при повышении температуры в «аномальной» области имеет необратимый характер. Изучено влияние начальной скорости замораживания на ход температурной зависимости.
В четвёртой главе изложены результаты исследования влияния отжига образца (при температурах из «аномальной» области) на концентрацию триплетных молекул акцептора энергии. Установлено, что отжиг раствора, так же как и повышение температуры в «аномальной» области, приводит к увеличению интенсивности фосфоресценции как молекул донора, так и сенсибилизированной фосфоресценции акцептора. Рост интенсивности сенсибилизированной фосфоресценции в процессе отжига обусловлен увеличением концентрации триплетных молекул акцептора энергии. На основании кинетических экспериментов показано, что относительная заселенность триплетного уровня молекул акцептора при этом практически не изменяется. Следовательно, увеличение числа триплетных молекул акцептора происходит за счет увеличения общего числа одиночных молекул в растворе, участвующих в излучении. На основании этого выдвинуто предположение, что увеличение числа одиночных молекул акцептора, участвующих в излучении сенсибилизированной фосфоресценции, в процессе отжига происходит в результате снятия концентрационного тушения. Исследована зависимость скорости увеличения числа триплетных молекул акцептора энергии от температуры отжига и определена энергия активации процесса (для различных донорно-акцепторных пар в различных растворителях).
Далее произведен анализ результатов работы. Рассмотрено влияние возможных механизмов концентрационного тушения в данных системах на параметры фосфоресценции донорно-акцепторной смеси и проведено сравнение их с экспериментальными данными. На основании этого сформулирован вывод о том, что основной вклад в увеличение интенсивности фосфоресценции молекул донорно-акцепторной смеси в результате отжига замороженного н.-парафинового раствора вносит распад гетероассоциатов. Это так же является причиной «аномального» характера температурной зависимости сенсибилизированной фосфоресценции.
В заключении суммированы основные результаты и выводы.
Научная новизна. Научная новизна работы заключается в том, что впервые исследовано влияние температуры на концентрацию триплетных молекул органических соединений в замороженных н.-парафиновых растворах при их сенсибилизированном возбуждении. Показано, что одной из причин концентрационного тушения триплетных молекул как акцептора, так и донора энергии в этих системах является образование ассоциатов при быстром замораживании раствора. Эти ассоциаты распадаются необратимо в процессе отжига образца, в результате чего тушение снимается.
Практическая и научная значимость. При разработке устройств, работающих на эффекте сенсибилизиации, часто возникает задача определения максимальных концентраций доноров энергии возбуждения и акцепторов, при которых достигается максимальный выход акцепторной люминесценции. Очевидно, что в отсутствие концентрационного тушения оптимальными являются максимально возможные концентрации как доноров, так и акцепторов. В реальных условиях всегда присутствуют различные процессы концентрационного тушения.
Результаты настоящей работы позволяют определить основные механизмы концентрационного тушения триплетных состояний органических молекул в условиях переноса энергии в кристаллических растворах. Обнаруженное влияние отжига на концентрацию одиночных молекул акцептора позволяет снимать концентрационное тушение триплетных состояний, тем самым целенаправленно повышая выход акцепторной люминесценции. Регулируя время и температуру отжига, можно руководить процессом увеличения концентрации одиночных молекул акцептора, что позволяет сделать процесс сенсибилизации в данных системах управляемым.
Обнаруженное действие отжига как процесса, приводящего к снятию концентрационного тушения в н.-парафиновых растворах, открывает перспективы исследования его применения к другим системам.
Результаты работы следует также учитывать при определении параметров переноса энергии в твердых растворах, так как характер тушения молекул донора акцепторами существенным образом зависит от предыстории образца.
Достоверность результатов. Достоверность представленных в диссертационной работе результатов обеспечивается: проведением опытов с использованием надёжных апробированных экспериментальных методик; совпадением определённых параметров фосфоресценции объектов исследования с литературными данными; согласованием между собой всех полученных результатов.
На защиту выносятся следующие положения:
1. Теоретическое обоснование методики определения концентрации триплетных молекул акцептора и константы перехода молекул акцептора из основного состояния в триплетное в результате переноса энергии из кинетических экспериментов.
2. Экспериментально обнаруженный немонотонный характер температурной зависимости интенсивности сенсибилизированной фосфоресценции органических молекул в н.-парафиновых растворах, причиной которого является снятие концентрационного тушения триплетных состояний в результате отжига раствора в процессе его нагревания.
3. Экспериментальное обоснование того, что увеличение концентрации триплетных молекул акцептора энергии в результате отжига происходит за счёт распада гетероассоциатов, которые вносят основной вклад в их концентрационное тушение.
4. Возможность использования отжига для увеличения эффективности протекания фотофизических процессов, происходящих с участием триплетных состояний органических молекул при их сенсибилизированном возбуждении.
Результаты исследований докладывались на:
- I-ой региональной научно-технической конференции «Вузовская наука – Северо-Кавказскому региону» г. Ставрополь, 14-16 мая 1997 г.;
- II-ой региональной научно-технической конференции «Вузовская наука – Северо-Кавказскому региону» г. Ставрополь, 1998 г.;
- III-ей региональной научно-технической конференции «Вузовская наука – Северо-Кавказскому региону» г. Ставрополь, 22-23 ноября 1999 г.;
- IV-ой региональной научно-технической конференции «Вузовская наука – Северо-Кавказскому региону» г. Ставрополь, 2000 г.;
- общефизических семинарах в Ставропольском государственном университете;
- научном семинаре в НИИ физики при Ростовском государственном университете, г. Ростов-на-Дону, январь 2000г;
- научном семинаре кафедры теоретической физики МПГУ им. Ленина,
г. Москва, июнь 2000г;
- Всероссийской научной конференции «Математическое моделирование в научных исследованиях» г. Ставрополь, 27-30 сентября 2000г.
По теме диссертации опубликованы работы [28-39].
Глава 1
Тушение возбужденных состояний примесных молекул в твердых растворах органических соединений. (Обзор литературы).
1.1 МЕЖМОЛЕКУЛЯРНЫЙ ПЕРЕНОС ЭНЕРГИИ ТРИПЛЕТНОГО ВОЗБУЖДЕНИЯ В ТВЁРДЫХ РАСТВОРАХ
Одним из распространенных механизмов дезактивации электронного возбуждения молекул является безызлучательный перенос энергии.
Безызлучательный перенос энергии электронного возбуждения представляет собой процесс, при котором возбуждённые молекулы донора энергии вступают во взаимодействие с невозбуждёнными молекулами акцептора энергии [19]. В результате такого взаимодействия появляется вероятность для перехода возбуждённой молекулы донора в электронно-колебательное состояние с меньшей энергией с одновременным переходом молекулы акцептора в состояние с большей энергией. В соответствии с законом сохранения энергии перенос энергии происходит только при условии, что спектры поглощения акцептора и спектры люминесценции донора перекрываются, т. е. в условиях резонанса.
Безызлучательный перенос энергии принято разделять на два вида: обменно-резонансный, когда перенос энергии осуществляется за счёт обменного взаимодействия, для возникновения которого необходимо перекрывание электронных облаков невозбуждённых молекул акцептора и возбуждённых молекул донора, и индуктивно-резонансный, когда электронные облака взаимодействующих молекул не перекрываются, а возбуждённые молекулы донора вступают в слабое кулоновское взаимодействие с невозбуждёнными молекулами акцептора.
Если электронные переходы в доноре и акцепторе разрешены правилами отбора, то перенос энергии происходит в результате диполь-дипольного взаимодействия. Для этого случая теория переноса энергии была развита Т. Фёрстером [40]. Она рассматривает процесс переноса энергии между молекулами в адиабатическом приближении и предполагает, что после переноса происходит быстрая колебательная релаксация в молекуле акцептора, что обеспечивает необратимость переноса энергии.
Вероятность переноса энергии в этом случае определяется из соотношения
![]() =
= 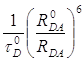 , (1)
, (1)
где ![]() - среднее время жизни
возбужденного состояния донора в отсутствии тушителя,
- среднее время жизни
возбужденного состояния донора в отсутствии тушителя, ![]() - расстояние между
молекулами,
- расстояние между
молекулами, ![]() - критическое расстояние
переноса (фёрстеровский радиус) – расстояние, на котором вероятность переноса
равна 1/
- критическое расстояние
переноса (фёрстеровский радиус) – расстояние, на котором вероятность переноса
равна 1/![]() . Величина
. Величина ![]() зависит от степени
перекрывания спектров донора и акцептора, а так же пропорциональна силам
осцилляторов переходов в доноре и акцепторе
зависит от степени
перекрывания спектров донора и акцептора, а так же пропорциональна силам
осцилляторов переходов в доноре и акцепторе
![]() ~
~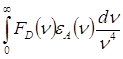 , (2)
, (2)
где ![]() - волновое число, FD(n) - квантовый спектр излучения донора, eА(n) - спектр поглощения акцептора; оба спектра нормированы на
единичную площадь.
- волновое число, FD(n) - квантовый спектр излучения донора, eА(n) - спектр поглощения акцептора; оба спектра нормированы на
единичную площадь.
Развитая Фёрстером теория явилась той основой, на которой базировалось дальнейшее изучение переноса энергии в случае обменных взаимодействий. Перенос энергии при обменном взаимодействии наблюдается, когда электронные переходы в акцепторе запрещены. В работе [41] Декстер Д. Л. показал, что в отличие от всех видов кулоновских взаимодействий, при обменных взаимодействиях константа переноса не зависит от силы осцилляторов переходов в доноре и акцепторе энергии, а зависимость её от расстояния в паре имеет экспоненциальный характер:
![]() ~
~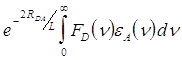 , (3)
, (3)
где L - средний эффективный боровский радиус возбуждённой молекулы донора и невозбуждённой акцептора.
Экспоненциальный множитель появляется вследствие того, что электронная плотность в молекуле, начиная с некоторой точки, спадает с расстоянием по экспоненте. Таким образом, при обменно-резонансных взаимодействиях вероятность переноса энергии уменьшается с увеличением расстояния между молекулами гораздо быстрее, чем в случае индуктивно-резонансных взаимодействий.
Переносом энергии при обменном взаимодействии объясняется сенсибилизированная фосфоресценция органических соединений. Условия, необходимые для переноса энергии, реализуются в основном в конденсированных средах. Впервые сенсибилизированная фосфоресценция была обнаружена в 1952 г. Терениным А. Н. и Ермолаевым В. Л. в твёрдых растворах органических соединений [42]. Они показали, что если в раствор нафталина добавить бензофенон или бензальдегид, то при облучении раствора светом ртутной лампы с длиной волны 365 нм наблюдается фосфоресценция нафталина, хотя нафталин излучение с данной длиной волны не поглощает. При повышении концентрации нафталина интенсивность фосфоресценции бензофенона уменьшается, а нафталина – увеличивается. Излучение нафталина было названо сенсибилизированной фосфоресценцией.
В последующих работах Ермолаева и Теренина [21,43] сенсибилизированная фосфоресценция наблюдалась для многих донорно-акцепторных пар в стеклообразных и кристаллических растворителях при низких температурах. Основные результаты можно сформулировать следующим образом. Спектр поглощения смеси представлен суммой полос поглощения донора и акцептора, дополнительных полос не наблюдается. Коротковолновые полосы спектра фосфоресценции доноров, не перекрываемые спектром сенсибилизированного свечения, не изменяются в присутствии акцептора. В спектрах и кинетике затухания фосфоресценции акцептора не наблюдается значительных отличий при прямом и сенсибилизированном возбуждении. Тушение донора экспоненциально растёт с увеличением концентрации акцептора. На основании этих результатов было сделано предположение, что сенсибилизированная фосфоресценция обусловлена переносом энергии триплетного возбуждения от молекул донора к молекулам акцептора, находящихся в основном состоянии. Результаты работ по изучению зависимости константы переноса от силы осциллятора перехода в акцепторе [21,44] показали, что изменение силы осциллятора перехода в акцепторе на четыре порядка не влечёт за собой заметного изменения вероятности переноса. Это позволило Ермолаеву и Теренину интерпретировать взаимодействия, обуславливающие триплет-триплетный перенос энергии, как обменно-резонансные.
К настоящему времени экспериментальные работы по безызлучательному триплет-триплетному переносу энергии в органических средах включают разные предметы исследования. Ведутся работы по изучению внутримолекулярного переноса энергии [45-50], переноса энергии в кристаллах [51-56], в твёрдых и жидких растворах [57-63], в тонких плёнках [64,65] и в газах [66,67].
В кристаллах физическая картина переноса энергии существенно отличается от других систем. Из-за трансляционной симметрии возможно возбуждение любой элементарной ячейки кристалла или же любой составляющих кристалл молекул. В этом случае перенос энергии обусловлен движением квазичастиц - экситонов. Экситонная теория переноса энергии в кристаллах заняла, судя по широкому кругу рассматриваемых интересов, самостоятельную область в разделе межмолекулярных взаимодействий.
В жидких растворах и газах перенос энергии электронного возбуждения контролируется диффузией. Диффузионные процессы также приводят к увеличению вероятности процессов, приводящих к безызлучательной дезактивации электронного возбуждения, за счёт чего фосфоресценция в жидкости затухает намного быстрее, чем в твёрдых растворах.
В твёрдых растворах молекулы находятся в триплетном состоянии более длительное время, поэтому представляется удобным исследовать основные характеристики межмолекулярного переноса энергии именно в данных системах. Исследования в данном направлении можно разделить по типу центров, между которыми наблюдается перенос энергии: а) между одиночными молекулами различных примесей, б) между одинаковыми молекулами примесей (миграция энергии), либо в) от основы (матрицы растворителя) к молекулам примеси.
При исследованиях переноса энергии между одиночными молекулами следует учитывать пространственное распределение молекул примесей в каждом из растворителей.
В стеклообразных растворителях, как и в жидкостях, в силу однородности
растворителя молекулы примеси равноудалены друг от друга. Функция распределения
молекул f(r) может
быть аппроксимирована d -
функцией, причём наиболее вероятное значение расстояний между ближайшими
молекулами ![]() определяется
средней концентрацией молекул
определяется
средней концентрацией молекул ![]() :
: ![]() ~
~![]() . Основным
условием переноса энергии между одиночными молекулами в данной среде является
хорошая растворимость молекул донора и акцептора для создания достаточно
высоких концентраций, при которых становится заметен безызлучательный
триплет-триплетный перенос энергии. Основные результаты по параметрам
межмолекулярного триплет-триплетного переноса энергии получены в основном в стеклообразных
растворах [7]. Зависимость константы скорости (3) от расстояния обычно
представляют в виде [10,68]
. Основным
условием переноса энергии между одиночными молекулами в данной среде является
хорошая растворимость молекул донора и акцептора для создания достаточно
высоких концентраций, при которых становится заметен безызлучательный
триплет-триплетный перенос энергии. Основные результаты по параметрам
межмолекулярного триплет-триплетного переноса энергии получены в основном в стеклообразных
растворах [7]. Зависимость константы скорости (3) от расстояния обычно
представляют в виде [10,68]
Kобм(r) = k0 exp [-2(r-R)/L], (4)
где k0 – константа скорости при контакте донора и акцептора, R – радиус запрещённого объёма. Для большого набора пар доноров и акцепторов триплетного возбуждения в ряде работ [10-14] определены критические расстояния переноса энергии R0 и сделана оценка параметров спада обменных взаимодействий L. Следует отметить, что даже для наиболее изученной пары бензофенон-нафталин получены параметры (табл. 1), очень сильно отличающиеся друг от друга.
Таблица 1
Параметры обменно-резонансного тушения фосфоресценции бензофенона нафталином в стеклообразных матрицах
|
R0, Å |
L, Å |
lg k0 |
Источник |
| 13,7 | 1,10 | 10 | [11] |
| 14,9 | 1,87 | 7,1 | [12] |
| 12,9 | 1,43 | 7,2 | [10] |
| 14,5 | 0,66 | 16 | [13] |
| 12,7 | 0,77 | 10,2 | [14] |
В работе [14] предполагается, что причиной подобного различия может быть некорректный анализ экспериментальных данных. Здесь же предложена экспериментальная методика и процедура обработки результатов, позволяющих корректно, по мнению авторов, извлекать информацию о механизме и параметрах тушения на основе анализа кинетических кривых затухания молекул донора в присутствии молекул акцептора.
Используя литературные значения где k0 и L можно рассчитать константу скорости динамического тушения в невязких жидкостях [69]:
kдин = (pL3/2) k0 (r 2+2r+2) » 2pR2Lk0, (5)
где r = 2R/L. Полученные значения kдин на 5-8 порядков ниже экспериментальных величин. Авторы работы [70] предполагают, что причиной этого может быть неэкспоненциальный характер зависимости константы скорости тушения от расстояния.
На наш взгляд, вышеуказанные несоответствия параметров переноса могут быть обусловлены и другими причинами. При определении параметров переноса считается, что тушение молекул донора обусловлено только одиночными молекулами акцептора, участвующими в излучении сенсибилизированной фосфоресценции. Однако при столь высоких концентрациях раствора в стеклообразных растворителях (10-2 – 1 М/л) не исключено появление нелюминесцирующих или слаболюминесцирующих ассоциатов различной степени сложности (как гомо-, так и гетероассоциатов), на которые происходит эффективный перенос энергии [19]. Это также может являться причиной некорректного определения k0 и L. Наличие такого механизма концентрационного тушения должно приводить к несоответствию параметров тушения фосфоресценции донора и сенсибилизированной фосфоресценции акцептора.
Следует отметить также, что в стеклообразных растворах даже при столь высокой растворимости, которую позволяют создать концентрации примесей 1 М/л, среднее расстояние в донорно-акцепторной паре составляет 12-15 Å. Поэтому можно считать, что теоретические выводы по обменно-резонансному механизму проверены только до определённых расстояний в донорно-акцепторной паре. Особенности же переноса энергии при более близком расположении молекул примесей до конца не изучены.
Взаимодействие молекул растворителя с молекулами активатора так же может вносить вклад в измеряемые параметры свечения.
В последние годы значительно возрос интерес к исследованиям взаимодействий между молекулами, адсорбируемыми на поверхности твёрдого тела [71-82]. Такие системы позволяют получить достаточно близкие расстояния между взаимодействующими молекулами, а так же интерес к таким системам обусловлен особенностями влияния микроскопической структуры матрицы на физические характеристики молекул.
Адсорбция примесей может быть получена на порошкообразных окисях магния и алюминия, на поверхности кремнозёма, в пористых и канальных матрицах. В качестве матриц используются стёкла, полученные по золь-гелевой технологии или путём выщелачивания натриевоборосиликатного стекла. Одна из особенностей заключается в том, что пространственное распределение молекул примесей носит фрактальный характер и характеризуется значительным разбросом расстояний между ближайшими соседними молекулами адсорбата, т.е. функция f(r) отличается от d - функции. Фракталы могут возникать либо в результате агрегации при диффузии (в них расстояние между ближайшими соседними частицами очень мало, постоянно и контролируется обычными, например ван-дер-ваальсовыми взаимодействиями между этими частицами), либо при взаимодействии с матрицей, вмещающей эти частицы [77].
Багничем С.А. исследовалась динамика триплетных возбуждений ароматических углеводородов при переходе от стеклообразных матриц к пористым [74,75]. Обнаружено отличие спектров фосфоресценции, кинетики затухания, и температурной зависимости интегральной интенсивности фосфоресценции бензофенона в этаноле, золь-гелевой матрице и пористом натриевоборосиликатном стекле. Спектр бензофенона в пористых матрицах смещается в коротковолновую область, затухание фосфоресценции носят неэкспоненциальный характер, причем времена затухания уменьшаются. Существенные различия наблюдаются в температурных зависимостях: в золь-гелевой матрице имеет место слабая зависимость интегральной по спектру интенсивности фосфоресценции, в натриевоборосиликатном стекле в области 77 -90 К происходит быстрое уменьшение интенсивности свечения. Обсуждая причины, лежащие в основе данных различий, автор говорит о наличии в пористых матрицах по крайней мере двух видов центров адсорбции, которые характеризуются противоположным действиям на фосфоресценцию карбонильных соединений. К одному из них можно отнести гидроксильные группы, структурно связанные с поверхностными атомами кремния, ко второму типу можно отнести поверхностные комплексы В(ОН)2+, сообщающие поверхности протонноакцепторные свойства [73].
Другими системами, в которых распределение молекул примесей существенно отличается от стеклообразных являются н.-парафиновые растворы. Н.-парафиновые растворители являются нейтральными по отношению к ароматическим углеводородам и поэтому оказывают меньшее влияние на параметры их люминесценции. Эти растворы кристаллизуются при замораживании, образуя снегообразную массу. При замораживании молекулы активатора могут либо внедряться в кристаллики растворителя по принципу замещения, либо вытесняться в межблочное пространство [25-27].
Ранее уже проводились исследования переноса энергии в данных системах [83,84]. Основной задачей авторов этих работ было получить квазилинейчатые спектры сенсибилизированной фосфоресценции. Однако получить квазилинейчатый спектр в условиях триплет-триплетного переноса энергии в таких системах долгое время не удавалось. Впервые наиболее узкий спектр сенсибилизированной фосфоресценции, принадлежащий индивидуальным молекулам акцептора , удалось получить для дифениленоксида в н.-гептане при 77К, используя в качестве донора антрон [85]. Далее были проведены исследования большого количества донорно-акцепторных пар в кристаллизующихся при 77К растворах и обнаружено, что только часть из них при определённой концентрации (10-3 моль/л) дают узкий спектр [86]. Однако, даже в случае самых узких линий они были шире, чем для обычной фосфоресценции акцептора. В качестве причины уширения спектров авторами была выдвинута гипотеза об увеличении силы электрон-фононной связи акцептора с матрицей в присутствии донора, но в дальнейшем она не получила полного экспериментального подтверждения.
Можно предположить, что причинами трудностей получения квазилинейчатых спектров в н.-прафиновых растворах является небольшая вероятность попарного внедрения молекул донора и акцептора. Большие используемые концентрации растворов предполагают не только внедрение, но и вытеснение молекул примесей на поверхность кристалликов, где на поверхности создаются повышенные локальные концентрации примесных молекул, а следовательно и благоприятные условия для переноса энергии. Люминесценция вытесненных молекул имеет диффузный характер и может быть интенсивнее квазилинейчатого спектра. Понижение же концентрации молекул примесей уменьшают вероятность попарного внедрения молекул донора и акцептора.
К настоящему времени остаётся нерешённым ещё ряд вопросов, связанных с триплет-триплетным переносом энергии электронного возбуждения.
Так, в работах Ермолаева В. Л. [7,21] показано, что квантовый выход сенсибилизированной фосфоресценции в отсутствие концентрационного тушения не зависит от концентрации раствора. Однако, как показали наши предварительные эксперименты [35], в н.-парафиновых растворах, уже начиная с концентраций примесей 10-3 М, во многих растворителях наблюдается падение квантового выхода сенсибилизированной фосфоресценции с увеличением концентрации молекул акцептора в растворе. По-видимому, это связано с агрегацией молекул примесей. Механизмы влияния агрегации на квантовый выход сенсибилизированной фосфоресценции к настоящему времени до конца не изучены.
В первых же работах по изучению триплет-триплетного переноса энергии не было обнаружено заметных различий между обычной и сенсибилизированной фосфоресценцией молекул примесей в стеклообразных растворах как в спектрах, так и в кинетике. В работе [87] было показано, что затухание сенсибилизированной фосфоресценции молекул аценафтена, дифенила и флуорена в замороженных растворах н.-гексана, толуола и этанола при 77 К происходит более быстро, чем обычной. Кроме того, в некоторых случаях наблюдался биэкспоненциальный характер кинетики. Причины такого различия к настоящему времени до конца не установлены. Следует заметить, что время затухания сенсибилизированной фосфоресценции тем больше отличается от затухания обычной фосфоресценции, чем больше концентрация молекул примеси в растворе. Поэтому не исключено, что и этот эффект каким-то образом связан с агрегацией молекул примесей.
Таким образом, в условиях межмолекулярного триплет-триплетного переноса энергии (сравнительно высокие концентрации) наряду с тушением триплетных молекул донора одиночными молекулами акцептора по-видимому возможны и другие механизмы тушения. Их наличие должно проявляться в несоответствии параметров тушения фосфоресценции донора и сенсибилизированной фосфоресценции акцептора. Поэтому для установления и изучения возможных механизмов тушения в условиях переноса энергии необходимо исследование наряду с параметрами фосфоресценции донора параметров сенсибилизированной фосфоресценции акцептора.
1.2 МЕХАНИЗМЫ КОНЦЕНТРАЦИОННОГО ТУШЕНИЯ ВОЗБУЖДЕННЫХ СОСТОЯНИЙ В РАСТВОРАХ
На тушение люминесценции при увеличении концентрации растворов обращал внимание ещё Вавилов [88]: «Свечение растворов (как и всякое свечение в обычной оптической трактовке) может быть характеризовано четырьмя свойствами – спектрами излучения и поглощения, выходом, поляризацией и длительностью. Опыт показывает, что все эти свойства при значительном возрастании концентрации раствора могут претерпевать изменения: спектры деформируются, выход падает, поляризация свечения так же убывает, уменьшается и длительность свечения».
Природа концентрационного тушения возбужденных состояний из-за сложности и многообразия возможных причин является и в наши дни предметом многочисленных исследований. В разное время этому явлению были даны различные объяснения, большинство из которых теперь имеют лишь исторический интерес.
В обзоре Южакова В. И. [20], обобщающем результаты экспериментальных и теоретических работ по концентрационному тушению возбужденных состояний, показано, что к тому времени наметились два основных подхода в объяснении его природы. Первый основывался на возможности индукционно-резонансной миграции электронного возбуждения между мономерными молекулами. Эти представления затем были положены в основу миграционной теории концентрационного тушения люминесценции. Согласно данной теории, тушение возбужденных состояний при больших концентрациях люминесцирующих веществ происходит за счёт резонансной передачи энергии электронного возбуждения от одной молекулы красителя, находящейся в мономерной форме к другой такой же молекуле. При этом часть таких переходов сопровождается тушением.
Другой подход в объяснении концентрационного тушения возбужденных состояний подчёркивал важность обратимой ассоциации молекул люминесцирующих веществ. Это явление объяснялось неактивным поглощением нелюминесцирующих ассоциатов. В дальнейшем была развита теория тушения люминесценции за счёт миграции энергии возбуждения с мономеров на ассоциаты. Ассоциационная теория концентрационного тушения возбужденных состояний, созданная Лёвшиным В. Л. с сотрудниками, предусматривает собственное неактивное поглощение ассоциатов и миграцию возбуждения с мономеров на эти ассоциаты.
В дальнейшем оба подхода были подтверждены последующими исследованиями.
Миграционная теория концентрационного тушения люминесценции была развита в ряде теоретических работ [8,15-17,89-94] и подтверждена экспериментально [18,71,72,95-98].
Под миграцией энергии подразумевается передача возбуждения только между центрами одинаковой природы. В зависимости от природы возбуждений их перенос осуществляется либо дальнодействующим (мультипольным), либо короткодействующим (обменным) межцентровым взаимодействием.
Делокализация возбуждения по системе случайно расположенных одинаковых центров складывается в диффузию. В работах [8,16,93] методами теории неупорядоченных систем найдена концентрационная зависимость коэффициента диффузии как при мультипольном, так и при обменном взаимодействии.
Однако возможность диффузии возбуждения на большие расстояния вовсе не означает, что его тушение обязательно является диффузионным. Зона тушения вокруг акцептора может быть настолько узка, что возбуждение способно попасть внутрь неё и выйти наружу однократным перемещением, а не последовательностью мелких шагов, складывающихся в континуальную диффузию. Одноактное тушение называют прыжковым. Скорости диффузионного и прыжкового тушения по разному зависят от концентрации доноров и микропараметров переноса возбуждения [8,16]. В разбавленных растворах, по мнению авторов [93], следует отдать предпочтение прыжковому механизму тушения.
В обзорах Бодунова Е. Н. [15,16] проведён анализ различных теоретических методов: Монте-Карло, непрерывных во времени случайных блужданий, эффективной среды и самосогласованный графический, используемых при исследовании спектральной миграции возбуждения в трёхмерных средах. Анализируется зависимость положения и формы неоднородно уширенного спектра люминесценции от времени и концентрации молекул при различных условиях возбуждения среды и механизмах межчастичного взаимодействия. Приводятся концентрационные зависимости квантового выхода люминесценции сред, содержащих два сорта молекул (доноров и акцепторов энергии возбуждений).
При вычислении параметров люминесценции в [15,16] основное внимание уделяется мультипольному взаимодействию. Для обменного взаимодействия вычисляется лишь коэффициент диффузии возбуждения. Это по-видимому связано с тем, что обменные взаимодействия осуществляются на меньших расстояниях по сравнению с мультипольными. Если индуктивно-резонансные взаимодействия разрешены правилами отбора, то они обладают преимуществом перед обменно-резонансными. Но если передача энергии по всем видам кулоновского взаимодействия запрещена, как в случае передачи энергии триплетного возбуждения, то обменный механизм миграции возбуждения является основным. Недостаток теоретических работ, рассматривающих влияние миграции триплетного возбуждения по системе случайно расположенных центров на параметры выхода их фосфоресценции, делает сложным выявление данного механизма тушения в рассматриваемых системах.
В работе [97] показано, что миграция энергии возбуждения в условиях неоднородного уширения спектров растворённого вещества при определённом соотношении между временами жизни возбуждённого состояния, миграции и релаксации приводит к концентрационному длинноволновому смещению спектров люминесценции органических красителей в различных растворителях. Как в твёрдых телах, так и в жидких растворах центры люминесценции одних и тех же веществ не являются идентичными вследствие различия их ближайшего окружения. При этом, помимо смещения энергетических уровней примесных центров, от величины локальных полей зависят и вероятности излучательных и безызлучательных переходов в молекулах растворенного вещества, а следовательно, и времена жизни возбуждённого состояния [77]. Направленность миграционных процессов при наличии расстройки энергетических уровней взаимодействующих молекул с повышением концентрации растворённого вещества приводит к увеличению заселённости первого возбуждённого состояния молекул с наиболее низко расположенными уровнями энергии. Направленная миграция на такие молекулы проявляется в концентрационном длинноволновом смещении спектров свечения. Если величина квантового выхода люминесценции молекул значительно уменьшается с понижением их возбуждённых уровней, то при этом также возникает концентрационное тушение люминесценции.
Однако, авторами [96] показано, что механизм миграции по мономерным молекулам обнаруживается только при отсутствии в растворе ассоциатов. Если в растворе имеются физико-химические образования, то изменения в спектрах и кинетике, обусловленные этими взаимодействиями превосходят остальные, упомянутые выше.
Увеличение концентрации раствора обычно сопровождается развитием межмолекулярных взаимодействий, часто приводящих к ассоциации молекул различной степени сложности. В результате в растворе наряду с мономерными молекулами появляются центры, существенно изменяющие оптические свойства раствора. Экспериментально наблюдаются разнообразные изменения спектров поглощения и люминесценции растворов, падение квантового выхода свечения и других параметров [99-103]. Это связано со сложными межмолекулярными взаимодействиями в растворах органических соединений и различной природой сил, объединяющих молекулы в ассоциаты.
Образование ассоциатов может происходить как за счёт сил Ван-дер-Ваальса, так и благодаря возникновению водородных связей [20,104]. Вклад ориентационного, индукционного и дисперсионного членов в Ван-дер-Ваальсовское взаимодействие определяется природой молекул примесей и растворителя.
Среди органических молекул наиболее изученными с точки зрения образования ассоциатов являются молекулы красителей и класса хлорофиллов [20]. Молекулы красителей хорошо ассоциируют в воде, в смесях полярных и неполярных растворителей. В полярных растворителях, где происходит сильная сольватация молекул красителей, их агрегация происходит при больших концентрациях. В неполярных растворителях молекулы хлорофилла ассоциируют уже при небольших концентрациях, однако красители в таких растворителях обычно не растворяются. Влияние растворителя на процессы ассоциации определяется тем, в какой степени способствует или препятствует объединению молекул красителей окружающая их сольватная оболочка.
Теория Ван-дер-Ваальсовых сил построена на предположении, что расстояние между взаимодействующими молекулами больше их поперечных размеров. При малых межмолекулярных расстояниях, характерных для высококонцентрированных растворов, вероятно, могут приобрести значение и силы, убывающие быстрее, чем 1/R6, например, силы, связанные с квадрупольным или обменным взаимодействием. В работе [105] было показано, что процессы ассоциации, вызывающие появление новых полос поглощения, характеризуют объединение молекул в одной общей сольватной оболочке на расстоянии ~ 8-10 Å. Обычно считается, что величина Ван-дер-Ваальсовского взаимодействия для молекул красителей не превышает 2 ккал/моль. Однако, это нельзя считать строго установленным фактом. В обзоре [20] упомянуто, что расчёты, выполненные Коулсоном и Девисом для молекул с мощными p-электронными облаками дают значение дисперсионного взаимодействия более десятка ккал/моль.
Другая точка зрения заключается в том, что объединение молекул красителей в ассоциаты происходит за счёт водородных связей. На основании анализа экспериментальных результатов, полученных в ряде работ для большого числа красителей, автором [19] показано, что представлению о дисперсионных силах, играющих основную роль при образовании ассоциатов, противоречат многие экспериментальные факты. Это прежде всего резкая зависимость эффективности процесса ассоциации от природы используемых растворителей и структуры молекул красителя. Наиболее подвержены образованию водородных связей полярные молекулы.
Образование ассоциатов может происходить и между различными молекулами. Разнородные ассоциаты обладают спектральными свойствами, отличными от мономеров и однородных ассоциатов. Они могут влиять на характер процессов переноса энергии возбуждения в смешанных растворах и служить дополнительными центрами её захвата.
Авторами [106] обнаружено существование как люминесцирующих разнородных ассоциатов – родамин 6Ж + метиленовый голубой, так и нелюминесцирующих – родамин 6Ж + бензопурпурин 4Б в буферных водных растворах. Здесь же показано, что перенос энергии на разнородные ассоциаты осуществляется с большей скоростью, чем на однородные или на мономеры. Так, критическое расстояние передачи энергии, рассчитанное авторами по спектральным характеристикам, между мономерами родамин 6Ж и метиленовый голубой R0 = 20 Ǻ, от мономерам к ассоциатам метиленового голубого R0 = 23 Ǻ, а от молекул родамин 6Ж на смешанные ассоциаты R0 = 47 Ǻ. Результаты говорят о том, что миграция на разнородные ассоциаты происходит со скоростью, большей почти на два порядка.
Как упоминалось выше, концентрационное тушение люминесценции вследствие неоднородного уширения спектров может быть обнаружено только при отсутствии в растворе более активных центров тушения. Таковыми являются либо другие примеси, эффективно осуществляющие тушение, либо однородные и разнородные ассоциаты. В этом случае миграция энергии возбуждения по системе одинаковых примесных центров, случайно, но равномерно распределёнными в твёрдой или жидкой среде, способствует доставке его к ловушкам энергии. С ростом концентрации донорных центров расстояние между ними сокращаются, а взаимодействие, ответственное за резонансный перенос энергии, усиливается. Это ускоряет миграцию возбуждения и сокращает время поиска ловушки, на которую энергия передаётся необратимо. В результате время жизни возбуждения монотонно сокращается с увеличением концентрации возбуждаемых центров [107]. Такой механизм тушения получил название миграционно-ускоренного тушения.
Процесс образования ассоциатов может осуществляться как между разнородными молекулами примесей, так и между молекулами примесей и растворителя. Например, в работе [108] исследованы молекулы аминокумаринов, которые обладают протонно-акцепторными свойствами. Взаимодействуя с молекулами протонно-донорного растворителя – этанола, они образуют комплексы с водородной связью. Н-связь возникает между карбонильной группой (>С=О) молекул красителей и оксигруппой (-ОН) растворителя. Вычислена энергия связи – 1400 см-1 . Такие комплексы обуславливают появление длинноволнового максимума в спектре поглощения и сдвиг максимума в спектре люминесценции (~35 нм при азотной температуре) в сторону коротких волн.
Таким образом, ассоциация молекул приводит к самым разным изменениям в спектрах поглощения и люминесценции молекул примесей. Это говорит о том, что вопрос о природе сил, объединяющих молекулы в ассоциаты, должен решаться в каждом конкретном случае по-своему. Силы же Ван-дер-Ваальса, как универсальные силы, всегда действуют между молекулами на достаточно близком расстоянии, однако в некоторых случаях теряют первостепенную роль.
В жидкости процесс ассоциации является обратимым. Все концентрационные эффекты полностью исчезают, а первоначальные оптические свойства растворов, содержащих мономерные молекулы исследуемого вещества, полностью восстанавливаются при обратном разведении концентрированных растворов [20].
Понижение температуры растворов сдвигает равновесие между мономерными молекулами и ассоциированными в сторону ассоциатов [20].
Кроме ассоциатов молекул в основном состоянии, возможно образование возбуждённых димеров. Они состоят из возбуждённой и невозбуждённой молекул, объединяющихся за время меньшее, чем средняя длительность их возбуждённого состояния. Эти возбуждённые димеры, получившие название эксимеров (одинаковые молекулы), эксиплексов (разные молекулы), обуславливают сильные изменения спектра люминесценции при постоянстве спектра поглощения [109-111].
Подводя итог анализа литературных данных по процессу ассоциации органических молекул, можно заметить, что основными объектами при его исследовании являлись жидкие растворы. Процессу образования ассоциатов в твердых растворах в литературе уделено значительно меньше внимания.
Обобщив наметившиеся к настоящему времени подходы к вопросу концентрационного тушения возбужденных состояний можно утверждать, что его причинами являются либо физико-химические (ФХ), либо резонансные (Р) взаимодействия между молекулами. Причём последние могут иметь обменный (близкодействующий), либо мультипольный (дальнодействующий) характер.
Эти причины могут приводить к изменению спектров поглощения и люминесценции, падению квантового выхода свечения, уменьшению времени его затухания. Основные механизмы, обуславливающие изменение параметров свечения в результате данных взаимодействий следующие:
1. Неактивное поглощение света ассоциатами (ФХ взаимодействие);
2. Миграция энергии между мономерными молекулами по индуктивно-резонансному или по обменно-резонансному механизму (Р взаимодействие);
3. Миграция энергии на ассоциаты (ФХ и Р взаимодействие);
4. Изменение внутримолекулярных безызлучательных констант. (ФХ и Р взаимодействие).
В литературе последний механизм тушения изучен недостаточно подробно, однако имеются факты, подтверждающие его существование и в ряде случаев его первостепенное значение. К одному из них можно отнести вопрос о том, почему образовавшиеся ассоциаты в основном не люминесцируют или слабо люминесцируют [77].
Рассмотренные механизмы могут обуславливать также тушение возбужденных состояний в условиях переноса энергии. Под таким углом зрения этот вопрос до настоящего времени не рассматривался. Хотя очевидно, что благоприятные для тушения условия создаются именно в концентрированных растворах, которые и являются необходимыми для наблюдения переноса энергии, особенно по обменно-резонансному механизму. Расхождения в определении параметров переноса энергии между органическими молекулами в твердых растворах ранее не связывались ни с возможным тушением, вызванным миграцией энергии, ни с влиянием ассоциатов на процесс перераспределения энергии в системе. Процесс ассоциации ароматических углеводородов так же остаётся малоизученным, тогда как именно на этих системах проверены основные параметры межмолекулярного триплет-триплетного переноса энергии.
На наличие дополнительных механизмов тушения, кроме переноса энергии с донора на акцептор, указывают также проведенные нами предварительные экспериментальные исследования. Они показали, что квантовый выход сенсибилизированной фосфоресценции может уменьшаться при увеличении концентрации молекул акцептора в растворе. Эти факты указывают на необходимость исследования механизмов тушения как в условиях прямого, так и сенсибилизированного возбуждения.
В случае однокомпонентных растворов ароматических углеводородов концентрационное поведение спектров излучения и поглощения в н.-парафиновых твёрдых растворителях исследовалось многими авторами (например, [26,112-115]).
Шпольским Э.В. с сотрудниками [26] были исследованы спектры флуоресценции и поглощения нафталина в н.-гептане и циклогексане при 77 К в диапазоне концентраций 10-2 – 10-4 М. Спектры флуоресценции растворов нафталина, состоящие при малых концентрациях из сравнительно широких полос, приобретают при увеличении концентрации квазилинейчатый характер. Авторы объясняют это следующим образом: « …за квазилинейчатые спектры флуоресценции и поглощения ответственны изолированные молекулы вещества "устроившиеся" в кристаллической матрице растворителя. Оказывается, что число таких молекул ограничено, так что для разных соединений существует предельная концентрация, выше которой интенсивность квазилинейчатого спектра не возрастает; если концентрация несколько превышает предельную, то на квазилинейчатый спектр флуоресценции накладывается полосатый молекулярный спектр, аналогичный спектру данного соединения в стеклообразном растворителе. Таким образом, за спектр флуоресценции растворов нафталина малых концентраций ответственны молекулы, "неустроенные" в кристаллической матрице растворителя. Повышение концентрации раствора приводит к агрегации таких молекул, чему способствует кристаллический характер матрицы. Агрегаты нафталина характеризуются своим собственным спектром поглощения и не люминесцируют. Поэтому при полной агрегации неустроенных молекул становится заметным квазилинейчатый спектр изолированных "устроившихся" в матрице растворителя молекул нафталина.»
Болотниковой Т. Н. и Наумовой Т. М. [116] установлено аналогичное поведение спектров фосфоресценции замороженных растворов нафталина в гексане и фенантрена в октане при изменении концентраций от 10-5 до 10-1М.
Таким образом, в н.-парафиновых растворах концентрационное тушение люминесценции наблюдается при более низких средних концентрациях, чем в стеклообразных матрицах. Это достигается созданием повышенных локальных концентраций молекул примесей на поверхностях микрокристаллов растворителя при вытеснении "лишних" молекул, превышающих предельную концентрацию "устроенных". Так как предел концентрации "устроенных" молекул определяется "удобством" растворителя, то в "неудобных" растворителях практически все молекулы примесей будут находиться на поверхности микрокристаллов. В таких условиях даже при низких концентрациях молекул примесей могут возникать условия, способствующие концентрационному тушению люминесценции, так же как и переносу энергии. Исследованию механизмов тушения люминесценции в "неудобных" н.-парафиновых растворителях в литературе уделено недостаточное внимание. Тем не менее, на наш взгляд, они являются хорошими модельными системами, позволяющими изучать особенности переноса энергии в условиях концентрационного тушения люминесценции.
1.3 ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА МЕЖМОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ ПРИМЕСНЫХ ОРГАНИЧЕСКИХ МОЛЕКУЛ В РАСТВОРАХ
Изучение влияния температуры на характеристики люминесценции растворов органолюминофоров являются одним из направлений исследования межмолекулярных взаимодействий в конденсированных средах.
По температурным зависимостям фосфоресценции примесных молекул в н.-парафиновых растворах можно определить их месторасположение в кристаллических матрицах [25,117-122].
Как упоминалось выше, в н.-парафинах молекулы растворённого вещества при кристаллизации могут оказаться внедрёнными в микрокристаллы парафина, либо могут быть вытеснены на поверхность. Молекулы примеси, внедрённые в микрокристаллы, жёстко фиксируются в них и хорошо изолированы друг от друга. Эти молекулы в случае слабого электрон-фононного взаимодействия дают квазилинейчатый спектр. Молекулы, адсорбированные на поверхности микрокристаллов, существуют либо в одиночном состоянии, либо в виде ассоциированных образований вплоть до агрегатов. Это зависит от концентрации примеси и «удобства» растворителя [25]. Для одиночных вытесненных молекул характерны сравнительно диффузные (хотя и достаточно структурные), полосатые молекулярные спектры, подобные таковым в стеклообразных растворителях. Ассоциация примесных молекул имеет преимущество в данном классе растворителей по сравнению со стеклообразными из-за создания их повышенных локальных концентраций при кристаллизации. В процессе роста микрокристаллов происходит вытеснение молекул примесей в оставшуюся область пространства. Агрегаты в ряде случаев характеризуются широкими бесструктурными полосами [25].
В работах Гребенщикова Д. М. И Персонова Р. И. [117-120] показано, что характер температурной зависимости фосфоресценции примесных молекул оказывается различным в случаях квазилинейчатого и диффузного спектров. Изучение температурной зависимости начальной интенсивности (I0) и длительности (t) фосфоресценции выявили два вида тушения: «низкотемпературное», начинающееся при 90 К и «высокотемпературное», процесс которого начинается вблизи точки плавления кристаллов. Это было продемонстрировано на примере поведения I0 и t фосфоресценции трифенилена в шести растворителях (от н.-гексана до ундекана).
От 77 до 90 К длительность фосфоресценции во всех растворителях практически не изменяется.
В области от 90 до 130 К характерна различная температурная зависимость I0 и t в разных н.-парафинах. В тех растворителях, где спектр обнаруживает хорошую квазилинейчатую структуру (н.-гексан и н.-гептан) существенные изменения I0 и t отсутствуют. В тех же растворителях, в которых спектр становится диффузным, с ростом температуры наблюдается резкое падение I0, сопровождающееся падением t (н.-декан и ундекан). Соответствующие зависимости для растворов в н.-октане и н.-нонане занимают промежуточное положение.
Резкое падение I0, сопровождающееся параллельным уменьшением t, свидетельствует о наличии эффективного тушения фосфоресценции, носящего диффузионный характер (тушение 2-го рода по Вавилову). В области сильного тушения кривые затухания могут быть представлены в виде суммы двух экспонент с различными показателями. При этом показатель одной из экспонент и её вклад в общую интенсивность с повышением температуры уменьшается, а показатель другой остаётся постоянным. При 150 К процесс тушения прекращается и время затухания снова принимает значение, близкое к t при 77 К. На основании этого авторы делают вывод, что в растворах, для спектров которых характерно наличие диффузных полос, фосфоресценция обусловлена двумя видами центров. Одни центры эффективно тушатся при Т > 90 К, а оставшееся слабое свечение принадлежит другим центрам, которые, как и в случае квазилинейчатых спектров, не испытывают «низкотемпературного» тушения.
В области от 130 К до точки плавления растворителя для всех растворов наблюдается «высокотемпературное» тушение – падение I0 и t.
Аналогичное поведение параметров фосфоресценции при изменении температуры наблюдалось для коронена, дифенилена, дифениленоксида в зависимости от удобства н.-парафинов [25].
На основании экспериментов по обезгаживанию растворов [117,118] установлено, что причиной уменьшения интенсивности и длительности фосфоресценции в обоих случаях является диффузионное кислородное тушение.
На основании данных исследований авторы делают следующие выводы. Центры, дающие диффузный спектр и испытывающие «низкотемпературное» тушение, образованы молекулами активатора, расположенными на поверхностях кристаллов парафина. Этим предположением легко объясняется тот факт, что начало тушения этих центров (90 К) практически совпадает с точкой кипения кислорода – процесс тушения в этом случае, по мнению авторов, осуществляется за счёт кислорода, находящегося в межкристаллических полостях, в порах и трещинах кристаллов. Центры, ответственные за квазилинейчатые спектры и испытывающие «высокотемпературное» тушение представляют собой молекулы активатора, внедрённые в кристаллики парафина. Их тушение обусловлено диффузией молекул кислорода внутри кристалликов парафина, и поэтому начало процесса тушения определённым образом зависит от свойств растворителя.
Таким образом, исследования параметров температурной зависимости позволяют судить о распределении примесных молекул в поликристаллических матрицах.
Как упоминалось выше, н.-парафиновые растворители использовались целым рядом исследователей для наблюдения межмолекулярного триплет-триплетного переноса энергии органических соединений в твердых растворах. Вопрос о местонахождении молекул примесей, участвующих в переносе энергии в данных системах к настоящему времени остается до конца невыясненным. Работы по температурной зависимости интенсивности и длительности обычной фосфоресценции примесных молекул дают основание предположить, что температурные исследования параметров сенсибилизированной фосфоресценции могут дать дополнительную информацию о месторасположении молекул примеси в н.-парафиновых матрицах, между которыми происходит эффективный перенос энергии электронного возбуждения.
Поскольку в «неудобных» растворителях молекулы примесей вытесняются на поверхность кристаллов, очевидным становится внимание к работам, посвящённым исследованию фотофизических процессов на поверхности твёрдого тела. В работе [123] изучены процессы триплет-триплетного переноса энергии и триплет-триплетной аннигиляции молекул красителей и ароматических углеводородов на поверхности кремнозёма. В температурных исследованиях свечения замедленной флуоресценции адсорбатов 1,2-безантрацена и антрацена в интервале температур 100-300 К обнаружено, что у адсорбатов ароматических углеводородов с условными моно- и полислоями имеет место экспоненциальный рост интенсивности с повышением температуры. Поверхность с малой домонослойной концентрацией ароматических углеводородов испускала замедленную флуоресценцию, которая не зависела от температуры и незначительно возрастала вблизи комнатных температур. Полученные результаты по обнаружению триплет-триплетной аннигиляции у ароматических углеводородов ещё домонослойных концентраций позволили авторам сделать вывод об островковой сорбции ароматических углеводородов. Это подтвердилось расчётом среднего расстояния между молекулами, равного 80 Å, в предположении о статическом распределении молекул, а также вычисленной константой скорости переноса триплетного возбуждения в этом случае из уравнения Штерна-Фольмера, которая меньше на два порядка литературных данных по тушению триплетных состояний. По мнению авторов при таких расстояниях перенос триплетной энергии и триплет-триплетная аннигиляция возможны только при условиях поверхностной диффузии. При этом триплет-триплетное взаимодействие в этих условиях осуществляется в контактных комплексах, которые образуются в результате миграции энергии по островку.
При больших концентрациях, когда образуется монослой из ароматических углеводородов, образование триплетных комплексов столкновения происходит по диффузионному механизму. Из наклона кривых замедленной флуоресценции в координатах ln(IЗФ0/IЗФ) от 1/Т были определены активационные барьеры температурного тушения замедленной флуоресценции, равные DЕa = 7.3 и DЕa = 9.3 кДж/моль для 1,2-бензантрацена и антрацена соответственно, которые авторы относят к процессам диффузионного перемещения физически сорбируемых молекул.
В работе Бегера В. Н., Земского В. И. [78] обнаружено, что температурные зависимости квантового выхода флуоресценции некоторых органических красителей в состоянии адсорбции носят существенно немонотонный характер. В диапазоне температур от 100 до 400 К имеются как интервалы уменьшения, так и увеличения (» от 110 до 200 К) квантового выхода флуоресценции. Авторами предложено объяснение выявленных особенностей способностью сложных молекул образовывать при адсорбции несколько устойчивых конфигураций относительно поверхности, отличающихся энергией взаимодействия адсорбат-адсорбент и эффективностью безызлучательной деградации энергии электронного возбуждения.
В ряде работ исследовано влияние температуры на эффективность передачи энергии триплетного возбуждения. Изменение передачи энергии от основы к примеси при понижении температуры исследовалось в [124,125]. Значительно меньше исследован вопрос о передаче энергии между самими примесными молекулами в кристаллах.
В работе Давыдова [126] показано, что учёт взаимодействия электронного возбуждения с колебаниями решётки приводит к зависимости вероятности передачи энергии от температуры.
В работе [127] экспериментально обнаружена передача энергии по триплетам между примесными молекулами фенантрена и нафталина в кристаллах дифенила, а так же уменьшение передачи при охлаждении до температуры жидкого азота.
В работе Жевандрова Н. Д. и Горшкова В. К. [128] исследовано влияние температуры на передачу энергии возбуждения синглетных состояний между молекулами разных примесей в кристаллах нафталина. Температурные тушения донора и акцептора энергии были учтены отдельно. Коэффициент передачи энергии вычислялся с учётом вероятности излучения – р, тушения - q и передачи - w:
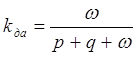 . (6)
. (6)
![]()
Значение
коэффициента передачи энергии получено из отношений интегральных интенсивностей
люминесценции и сенсибилизированной люминесценции для двух значений
температуры. Полученные значения коэффициентов передачи при температуре паров
жидкого гелия и при комнатной равны соответственно для пары антрацен-нафтацен kдагел = 0.30 и kдаком = 0.63, а для
пары антрацен-люмоген kдагел = 0.45 и kдаком = 0.72. Следовательно,
при понижении температуры до 6 К коэффициент передачи энергии между примесными
молекулами в кристаллах нафталина уменьшается примерно в 2 раза.
Тот факт, что в полистирольных плёнках для этих же пар не наблюдалось изменение передачи энергии при изменении температуры [129], связан, по мнению авторов, с отсутствием кристаллической структуры. В некристаллической матрице все положения молекул равноценны, и, с учётом теории Давыдова, при возбуждении не происходит возникновение новых равновесных положений и новых колебательных состояний молекул. Поэтому зависимость формы полос от температуры выражена слабо, что объясняет отсутствие заметных изменений передачи энергии с температурой между молекулами донора и акцептора в некристаллической матрице, тогда как для кристаллической эти изменения весьма существенны.
В кристаллических матрицах величина взаимодействия электронного возбуждения с колебаниями решётки должна определяться характером этого взаимодействия. Поэтому зависимость коэффициента передачи энергии между примесными молекулами может сильно изменяться в зависимости от месторасположения молекул примеси в кристаллической матрице и характера их взаимодействия.
В н.-парафиновых твёрдых растворах влияние температуры на эффективность переноса энергии между молекулами различных примесей не исследовалось.
Опираясь на результаты температурных исследований, можно разделять виды тушения люминесценции. Согласно классификации, предложенной С.И. Вавиловым, виды тушения делятся на тушения первого и второго рода [130]. Тушением первого рода названы процессы, в которых выход люминесценции уменьшается при воздействиях на невозбуждённые молекулы вещества (такое тушение так же носит название статического [19]). Под тушением второго рода понимают процессы, в которых выход люминесценции уменьшается при воздействии на возбуждённые молекулы вещества.
Примером статического тушения может служить образование нефлуоресцирующих комплексов в основном состоянии, примером тушения второго рода – динамическое тушение, заключающееся в переносе энергии на невозбуждённые молекулы примеси. При тушении первого рода средняя длительность t возбуждённого состояния молекул сохраняется постоянной, так как в возбуждённое состояние переходят лишь те из них, которые избежали внешних воздействий; при тушении второго рода из-за воздействия на возбуждённые молекулы вещества t изменяется.
Для числа молекул в возбуждённом состоянии можно записать следующее выражение:
nВ = b N, (7)
где N – общее число молекул, b - коэффициент, показывающий, какая часть от общего числа молекул находится в возбуждённом состоянии.
Учитывая вышеуказанные типы тушения, можно сказать, что статическое тушение возбуждённых состояний обусловлено уменьшением N, а динамическое – уменьшением b.
Влияние температуры на статическое и динамическое тушение различно. Если статическое тушение обусловлено существованием ассоциатов, то повышение температуры уменьшает их стабильность, тем самым увеличивая общее число одиночных молекул N в растворе. Это ведёт за собой увеличение числа возбуждённых молекул и как следствие, увеличение интенсивности люминесценции.
Константа же динамического тушения с ростом температуры увеличивается [19]. Примером динамического тушения может служить кислородное тушение люминесценции в твёрдых растворах. Константа динамического тушения определяется скоростью, с которой молекулы кислорода диффундируют к центрам взаимодействия, а коэффициент диффузии увеличивается с ростом температуры.
Примером влияния температуры на статическое тушение люминесценции могут служить результаты работы [106]. Так, например, относительный выход люминесценции при нагревании водного раствора родамина 6Ж и бензопурпурина 4Б от 20 до 50° С увеличивается ~ в 2,7 раза. Т.е. он практически восстанавливается до значения, с которого начинается падение выхода свечения родамина 6Ж при увеличении содержания в растворе бензопурпурина 4Б. Это указывает на разрушение смешанных ассоциатов при нагревании.
Исследования Сапунова В.В. и Егоровой Г.Д. процесса ассоциации ряда порфиринов в водных растворителях выявили Аррениусовскую зависимость константы образования ассоциатов от температуры [129]:
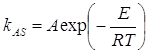 , (8)
, (8)
где Е – энергия активации рассматриваемого процесса.
Основная часть работ по изучению влияния температуры на степень ассоциации выполнена для жидких растворов. Подобных работ для твёрдых растворов в литературе обнаружить не удалось. В жидких растворах увеличение числа мономерных молекул при повышении температуры имеет обратимый характер, т.е. при понижении температуры их число уменьшается. По нашему мнению, в твёрдых растворах повышение температуры по той же причине, как и в жидких растворах, должно приводить к увеличению доли мономерных молекул. Однако, в этом случае может произойти после распада нарушение условий, необходимых для образования ассоциатов (изменение среднего расстояния между молекулами, их взаимной ориентации и т.д.). Такие различия могут быть обусловлены различной скоростью диффузионных процессов в жидкости и в твёрдом теле.
Одной из задач раздела химии, изучающего макрокинетику, является рассмотрение роли диффузии в химических процессах. «Как правило, результирующая скорость процессов в реальных системах определяется совместным переносом реагирующих между собой компонентов» [131]. Образование и распад ассоциатов, подобно химическим реакциям, обусловлены процессами межчастчного взаимодействия, только с меньшей энергией активации, и так же подчиняются законам химической кинетики. Вполне очевидно, что процессы межмолекулярного взаимодействия в реальных системах также должны быть взаимосвязаны с процессами переноса и описываться законами макрокинетики.
Для диффузии примесных молекул и процесса ассоциации, составляющих в совокупности реальный процесс, существуют характерные времена протекания – соответственно tD и tAS. Если tD « tAS, наблюдаемая скорость реакции должна определяться законами химической кинетики. В этом случае можно говорить о кинетическом режиме реакции. Если tD »tAS или это величины одного порядка, наблюдаемая скорость реакции в той или иной степени должна определяться скоростью диффузии реагентов. В этом случае можно говорить о диффузионном режиме реакции.
Для молекулярной диффузии tD = L2/D, где L – пространственная область, характеризующая её протекание, D – коэффициент диффузии. В случае рассмотрения влияния диффузии на процесс ассоциации, L – область взаимодействий, обуславливающих межмолекулярную связь.
Характер зависимости коэффициента диффузии от температуры в жидкости и в твёрдом теле существенно различен. В жидкости
D = kT/6prh, (9)
где r – размеры молекул, h - вязкость жидкости. В твёрдом теле
D = D0 ехр(-ED / kT), (10)
где D0 – фактор диффузии, ED – энергия активации диффузии.
Соответственно, tD ~ h/Т в жидкости, tD ~ ехр (ЕD /kT) в твёрдом теле. Таким образом, при фазовом переходе растворителя твёрдое тело – жидкость наблюдается изменение характера зависимости tD от температуры.
Согласно законам химической кинетики скорость реакций обычно описывается Аррениусовской зависимостью константы скорости процесса q от температуры:
q(Т) ~ ехр (-ЕAS/kT) (11)
где ЕAS- энергия активации процесса.
Соответственно для tAS:
tAS ~ ехр (ЕAS/kT). (12)
Вид зависимости определяется непосредственно характером наблюдаемого взаимодействия частиц в ассоциате и не зависит от агрегатного состояния растворителя. Таким образом, основываясь на данных рассуждениях, можно предположить, что при переходе от твёрдого тела к жидкости не происходит резкого изменения хода кривой tAS(T ).
Экспоненциальная зависимость t от 1/T предполагает резкое падение t в начальной области и стремление к постоянному минимальному значению t∞ при высоких температурах как в случае диффузии, так и в процессе ассоциации. Знание коэффициентов ЕAS, ED и tАS∞ и tD∞ могут позволить качественно оценить режим процесса ассоциации в твёрдом теле.
Представленный рисунок 1 качественно отражает изменение характерных времён процессов при повышении температуры. Прямая tD ~ h/Т изображена условно, без учёта зависимости h(Т). При рассмотрении н.-парафинов точка плавления соответствует энергии kT, равной 1-2 кДж/моль. Энергия связи в ассоциатах органических молекул лежит в широком интервале – от единиц до десятков кДж/моль. Однако даже этой грубой оценки оказывается достаточно, чтобы увидеть, что в твёрдой фазе н.-парафинов наблюдается резкое падение кривой tAS(Т). Ход кривой tD(Т) в твёрдой фазе трудно предсказать из-за отсутствия данных по порядку величины ED примесных органических молекул в замороженных н.-парафиновых матрицах. Точка пересечения, изображённая на графике в жидкой фазе и соответствующая равенству tD и tAS может находиться, скорей всего и в твёрдой фазе. Этот вопрос требует дальнейшего исследования.

В
жидкости при высоких температурах (kT > ЕAS) преимущественно должен преобладать диффузионный режим процесса ассоциации. На кривой tAS(T ) (см. рис.1) эта область характеризуется медленным падением tAS. А так как в жидкости наблюдается падение tD с постоянной скоростью, то в этой области температур tD » tAS, что приводит к диффузионному режиму реакции.
Эксперимент свидетельствует, что при неизменной температуре в жидкости устанавливается равновесие в системе: число образовавшихся ассоциатов равно числу распавшихся. Повышение температуры приводит к смещению равновесия в сторону мономерных молекул. Если рассмотреть согласование эксперимента с макрокинетическим подходом в жидкости, то при повышении температуры в диффузионном режиме время «оседлого состояния» молекул становится меньше характерного времени образования ассоциата и вероятность образования ассоциатов при этом уменьшается, что согласуется с экспериментальными данными.
В литературе данных по макрокинетическому подходу к процессу образования ассоциатов не было обнаружено. Изучение этого вопроса в твёрдом теле представляет интерес с точки зрения возможного кинетического режима реакции. Для примесных молекул в твёрдых растворах процесс образования ассоциатов может быть затруднён из-за малой вероятности попадания молекул в процессе диффузии в зону взаимодействий L (при tD « tAS). Межмолекулярное расстояние после распада ассоциата из-за несимметричности потенциальной кривой взаимодействия больше, чем между взаимодействующими молекулами. Соответственно, сразу после распада условия образования ассоциата могут быть нарушены. Далее, «медленный» в этом случае процесс диффузии молекул идёт в сторону наименьшей концентрации, т.е. способствует удалению молекул друг от друга. В этом случае образования ассоциатов наблюдаться практически не будет. А следовательно, процесс распада ассоциатов в этих условиях должен быть необратимым. Высказанные предположения о процессах ассоциации примесных молекул в твёрдых растворах требуют дальнейших подтверждений.
Подводя итог вышесказанному, можно предположить, что температурные исследования в условиях триплет-триплетного переноса энергии открывают возможность получения многосторонней информации о межмолекулярных взаимодействиях в н.-парафиновых матрицах. Во-первых, по характеру спектров и ходу температурной зависимости относительной интенсивности обычной и сенсибилизированной фосфоресценции можно будет судить о месторасположении в кристаллах молекул, участвующих в переносе энергии. Во-вторых, характер температурной зависимости относительной интенсивности и длительности фосфоресценции донора и сенсибилизированной фосфоресценции акцептора может определить основные механизмы тушения в данных системах. В-третьих, кинетические эксперименты позволят контролировать число одиночных молекул при различных температурах (описано ниже), что открывает возможности исследования влияния различных процессов на концентрационное тушение возбужденных состояний.
Таким образом, анализ литературных данных показал, что к настоящему времени остался нерешенным вопрос о механизмах концентрационного тушения в условиях межмолекулярного триплет-триплетного переноса энергии в твердых растворах. Настоящая работа посвящена исследованию влияния температуры на параметры фосфоресценции донора и сенсибилизированной фосфоресценции акцептора в замороженных н.-парафиновых растворах органических соединений и установлению механизмов этого влияния. Это позволит получить информацию о существующих механизмах тушения в условиях межмолекулярного триплет-триплетного переноса энергии.
Глава 2
Методика экспериментальных исследований
В данной главе рассмотрены методические вопросы. Обосновывается выбор объектов исследования. Описывается экспериментальная установка, методика спектральных, кинетических и температурных измерений. Наряду с этим, решаются задачи определения концентрации триплетных молекул акцептора энергии, константы перехода молекул акцептора в триплетное состояние из изучения кинетики их накопления и распада. Решение этих задач необходимо для последующих исследований влияния температуры на концентрацию молекул акцептора энергии в триплетном состоянии.
2.1 РАСТВОРИТЕЛИ И СОЕДИНЕНИЯ
Как было показано в главе 1, в н.-парафинах локальная концентрация примеси может быть намного больше, чем средняя концентрация её в стеклообразных растворах при тех же условиях. Благодаря этому представляется возможность исследовать особенности переноса энергии при меньших расстояниях между молекулами в донорно-акцепторной паре. По этой же причине в данных системах так же должны быть более выражены механизмы концентрационного тушения триплетных состояний, вследствие чего они становятся более доступными для экспериментальных исследований. Этим и высокой растворимостью ароматических углеводородов в некоторых н.-парафинах обусловлен их выбор в качестве растворителей.
Для решения поставленной задачи был выбран ряд н.-парафинов: от н.-гексана до н.-декана. Как показал проведенный анализ литературных данных, в н.-гексане и н.-гептане рассматриваемые молекулы примесей могут как внедряться в матрицы растворителя, так и находиться на поверхности. Н.-октан и н.-декан являются «неудобными» матрицами для доноров и акцепторов энергии, и при замораживании молекулы примесей преимущественно вытесняются на поверхность. Н-нонан был исключён из используемых растворителей, ввиду наличия фазового перехода второго рода в исследуемом интервале температур (от 77 до 122 К).
Точки плавления н.-парафинов представлены в табл. 2.
Таблица 2
Точки плавления растворителей
| Растворитель | Точка плавления, К |
| н.-гексан | 178 |
| н.-гептан | 182 |
| н.-октан | 216 |
| н.-декан | 243 |
Растворители – н.-гексан и н.-октан марки «ч», н.-гептан и н.-декан марки «хч», дополнительно очищались двукратной фракционной перегонкой. Отсутствие фосфоресценции растворителя в условиях проведения эксперимента являлось критерием его чистоты.
При выборе объектов исследования необходимо было учесть требования, которым должны удовлетворять молекулы донорно-акцепторной пары [7]:
1. Триплетный уровень молекул донора энергии должен быть расположен выше триплетного уровня молекул акцептора (закон сохранения энергии).
2. Для осуществления избирательного возбуждения только молекул донора энергии их флуоресцентный уровень должен быть ниже соответствующего уровня молекул акцептора.
При выполнении этих условий синглет- синглетный перенос энергии невозможен из-за неблагоприятного расположения энергетических уровней, а триплет-триплетный перенос наблюдается, если молекулы находятся в радиусе обменных взаимодействий.
Однако, выполнение первого условия ведет к перекрыванию спектров обычной фосфоресценции донора и сенсибилизированной фосфоресценции молекул акцептора. Разделить их спектрально в большинстве случаев представляется достаточно сложно. Поэтому для удобства экспериментальных исследований необходимо, чтобы время жизни молекул акцептора в триплетном состоянии превышало время жизни триплетных молекул донора на несколько порядков. Это позволяет отделить сенсибилизированную фосфоресценцию молекул акцептора от фосфоресценции донора во времени.
Если, например, взять в качестве акцептора молекулы, время жизни которых в триплетном состоянии несколько секунд, а в качестве донора – молекулы, время жизни которых в триплетном состоянии несколько миллисекунд, то уже спустя 0.1 - 0.5 с после прекращения возбуждения свечение полностью определяется фосфоресценцией акцептора.
С учётом вышеперечисленных ограничений в качестве доноров энергии были выбраны ароматические кетоны: бензофенон и антрон, квантовый выход триплетных состояний которых близок к единице [132]. В качестве акцепторов – нафталин, флуорен и аценафтен.
Фосфоресценция этих соединений при низких температурах достаточно хорошо изучена. Основные спектральные и люминесцентные характеристики исследуемых ароматических углеводородов представлены в табл. 3. Как видно из приведённых данных, для любой комбинации выбранных доноров и акцепторов энергии выполняются вышеуказанные требования к донорно-акцепторным парам.
Таблица 3
Основные характеристики используемых соединений
| Соединение | Молекуляр-ная формула | Растворитель |
Т-уровень, см-1 |
S1-уровень, см-1 |
tфосф, с |
Источник |
Доноры |
С13Н10О |
Смесь этанола и диэтилового эфира |
24250 | 26000 |
4.7×10-3 |
[7] |
Бензофенон |
||||||
Антрон |
С14Н10О |
Смесь этанола и диэтилового эфира |
25150 | 27000 |
1.5×10-3 |
[7] |
АкцепторыС10Н8 |
Смесь этанола и диэтилового эфира |
21250 | 31750 | 2.3 | [7] | |
| Нафталин | ||||||
Аценафтен |
С12Н10 |
н.- гексан н.-гексан |
20870 - |
31180 - |
- 2.82 |
[116] [87] |
Флуорен |
С13Н10 |
н.-гептан | 23760 | 33220 | 6.15 | [87] |
БЕНЗОФЕНОН. Фосфоресценция бензофенона изучалась рядом авторов как для различных кристаллических модификаций [133-136], так и для твёрдых растворов.
В твёрдых растворах в зависимости от характера взаимодействия с молекулами растворителя наблюдаются изменение положения и структуры электронных спектров, квантового выхода и длительности излучения молекул активатора.
В стеклообразных растворителях (спиртово-эфирные смеси) [7] спектры фосфоресценции ароматических углеводородов имеют диффузный характер.
Гаевским А.С. с соавторами [137] исследовались спектры фосфоресценции кристаллического, стеклообразного бензофенона и его раствора в спитово-эфирной смеси при температурах 90 К. Авторами показано, что максимум и ширина 0-0 полосы, а так же время затухания фосфоресценции бензофенона в каждом состоянии различные (табл. 4).
Таблица 4
Характеристики излучения бензофенона в зависимости от фазового состояния
| Состояние |
Положение максимума 0-0 полосы, (lмах, нм) |
Полуширина, (Dn, см-1) |
Время затухания фосфорсценции,(t, с) |
| Раствор | 414 | 700 |
4.7×10-3 |
| Кристаллический | 416 | 300-600 |
» 7×10-4 |
| Стеклообразный | 427 | 700 |
3.4×10-3 |
Авторами [138] показано, что интенсивность и время жизни фосфоресценции бензофенона в матрицах полиметилметакрилата меняется при фазовых переходах в растворителе.
В пористой золь-гелевой матрице и в пористом натриевоборосиликатном стекле [73-75] спектр бензофенона уширяется по сравнению с этанольным раствором, наблюдается его смещение в коротковолновую область (порядка 10 нм), а так же уменьшение времени затухания и изменения в температурной зависимости.
Авторами [14] исследованы кинетические кривые затухания фосфоресценции бензофенона в различных растворителях. Получены значения времени затухания фосфоресценции бензофенона в этаноле – (6,2 ± 0,2) мс, в смеси этанол-эфир (2:1) – (5,4 ± 0,2) мс и в смеси эфир-метилциклогексан (2:1) – (5 ± 0,2) мс.
Таким образом, фосфоресценция бензофенона испытывает существенные изменения в зависимости от растворителя и условий её наблюдения.
Авторами работы [86] была исследована фосфоресценция бензофенона в н.-гексане и н.-гептане и показано, что спектр в данных растворителях имеет диффузный характер.
 |
В диссертационном исследовании, непосредственно перед опытами, где бензофенон использовался в качестве донора энергии, была подробно изучена его фосфоресценция в каждом из растворителей. В качестве примера приведён спектр фосфоресценции бензофенона в н.-октане (рис. 2). Максимуму 0-0 полосы в спектре фосфоресценции соответствует длина волны - 418 нм, полуширина полосы – 650 см-1. Время затухания фосфоресценции бензофенона – 7 мс. Сравнивая полученные данные с результатами исследований, представленных в табл. 4, можно сделать заключение, что они ближе всего к люминесценции раствора бензофенона. К кристаллическому бензофенону их нельзя отнести, поскольку время затухания фосфоресценции кристаллического бензофенона на порядок меньше наблюдаемого, а так же ширина полос кристаллического бензофенона меньше наблюдаемой. По сравнению со стеклообразным бензофеноном наблюдается значительное расхождение в положении максимума. Поэтому спектр был отнесён к фосфоресценции мономерных молекул бензофенона, однако в н.-парафиновых растворах наблюдается длинноволновое смещение максимума на 4-6 нм по сравнению со стеклообразными растворителями, что свидетельствует, по всей вероятности, о создании повышенных локальных концентраций примесей.
Бензофенон так же использовался в качестве донора энергии в целом ряде работ как в стеклообразных [7,14], так и в н.-парафиновых [86] растворителях.
АНТРОН. Антрон относится к типу молекул, обладающих короткоживущей фосфоресценцией с высоким квантовым выходом. Спектры антрона в н.-парафиновых растворах от н.-пентана до н.-декана при 77К изучены Гобовым Г.В. и Конашенко В.И. [139]. В широком диапазоне концентраций были получены квазилинейчатые спектры от н.-пентана до н.-октана, имеющие мультиплетную структуру с различным числом компонент.
Этими же авторами антрон использовался в качестве донора энергии при исследовании сенсибилизированной фосфоресценции органических молекул в н.-парафиновых растворах [140].
НАФТАЛИН. Спектры флуоресценции нафталина в н.-парафинах при 77К были впервые изучены Болотниковой [113,114]. Наблюдалось два вида спектров. Хорошо разрешенный квазилинейчатый спектр нафталина наблюдался в н.-пентане и циклогесане. В других н.-парафинах в пределах концентраций, при которых проводились исследования (10-3-10-4 моль/л) спектр терял квазилинейчатую структуру и состоял из размытых полос, как в стеклообразном растворе.
Dekkers J. J. с соавторами [115] исследовались спектры флуоресценции нафталина в н-парафинах при 20 К. При концентрации примеси 10-4 М получены диффузные молекулярные полосы от н.-гексана до н.-октана и только в случае н.-пентана наблюдаются резкие структурные линии.
Донорно-акцепторная пара бензофенон-нафталин наиболее часто использовалась в качестве объекта исследований при изучении триплет-триплетного переноса энергии [7,14,42,70]. В [7] приведены значения квантового выхода обычной фосфоресценции нафталина » 0,05 (в смеси эфира, изопентана и спирта) и сенсибилизированной фосфоресценции » 0,06 - 0,07 (в спиртово-эфирной смеси) при 77 К. Спектр сенсибилизированной фосфоресценции нафталина в н.-гексане, донор – бензофенон, как и в стеклообразных растворах, представляет собой молекулярные полосы [86].
Образованию эксимеров, эксиплексов, агрегированных комплексов с участием молекул нафталина посвящен целый ряд работ [141-146].
АЦЕНАФТЕН. Спектры флуоресценции и фосфоресценции аценафтена в н.-парафинах изучены достаточно подробно при различных концентрациях примеси Мамедовым Х. И. [147] и Dekkers J. J. [148]. Как и для остальных рассматриваемых соединений вид спектра люминесценции аценафтена зависит от подбора растворителя и концентрации примеси.
Наиболее удобным растворителем для аценафтена является н.-пентан, в котором спектр люминесценции в широком диапазоне конценцтраций (10-5-10-2 М) является квазилинейчатым [148]. При дальнейшем увеличении длины цепочки растворителя для малых концентраций спектр преобразуется в диффузные полосы. Так, в н.-гексане такой предельной концентрацией является 10-5 М , в н.-гептане –10-4 М, а в н.-октане квазилиний не наблюдается вообще. Представляет интерес уменьшение интенсивности свечения в последнем растворителе приблизительно на порядок при увеличении концентрации от 10-4 М до 10-2 М. Максимум диффузной полосы при этом немного смещается в коротковолновую область, а в длинноволновой области, отстоящей более чем на 1000 см-1, появляется широкое диффузное свечение, принадлежащее кристаллическому аценафтену.
При исследовании концентрационной зависимости спектров фосфоресценции аценафтена в матрицах н.-гексана при 77 К [149] наблюдалось три типа молекулярных спектров. Для концентраций раствора от 10-2 М до10-4 М наблюдался квазилинейчатый спектр. Для концентраций раствора, меньших чем 10-3 М, в спектре с коротковолновой стороны от квазилиний наблюдались широкие молекулярные полосы, смещенные на 50 см-1 и подобные полосам в спектре флуоресценции при тех же концентрациях. В узком интервале концентраций в области 10-1 М наряду с квазилиниями появились полосы, смещенные в длинноволновую область спектра относительно квазилиний на 200 см-1. На основании результатов температурной зависимости спектров фосфоресценции аценафтена авторами выдвинуто предположение, что за первый тип центров отвечают молекулы, внедренные в кристаллы, за второй – одиночные молекулы аценафтена, вытесненные на поверхность. Третий тип обусловлен свечением центров, внедренных в кристаллы н.-гексана, однако большая ширина и их смещение, по-видимому, связаны с неоднородным уширением и увеличением электрон-фононного взаимодействия из-за высоких концентраций.
В работе [28] был исследован спектр и кинетика сенсибилизированной фосфоресценции аценафтена в кристаллическом бензофеноне при переносе энергии от основы к примеси. Работа была выполнена с целью исключить из рассмотрения данный тип центров (микрокристаллы донора с внедренными в них молекулами акцептора) при условиях создания больших концентраций примеси (10-1 М). Как показал проведенный анализ, максимум 0-0 полосы сенсибилизированной фосфоресценции смещен на 120 см-1 в длинноволновую область по отношению к максимуму этой же полосы в н.-гексане. Полуширина 0-0 полосы сенсибилизированной фосфоресценции аценафтена в кристаллическом бензофеноне составляет около 240 см-1. Среднее значение времени затухания для интегральной ( без разложения в спектр) интенсивности составляет 2.40 с, что заметно отличается от среднего времени затухания сенсибилизированной фосфоресценции аценафтена в н.-гексане (табл. 2).
Достоверные тонкоструктурные спектры аценафтена в основном и возбужденном электронных состояниях, не искаженные влиянием на молекулы окружающей среды, получены в [150] при охлаждении в сверхзвуковой струе.
ФЛУОРЕН. Исследование люминесцентных характеристик флуорена было произведено преимущественно в условиях эффекта Шпольского. Квазилинейчатые спектры флуорена изучены в работах Нурмухаметова Р. Н. и Гобова Г. В. с соавторами [151,152]. Наиболее отчетливая структура в спектрах флуоресценции и фосфоресценции получена в н.-гексане и н.-гептане. Фосфоресценция по интенсивности сравнима с флуоресценцией.
Гобовым Г. В. и Конашенко В. И. [140] были получены наиболее узкие спектры сенсибилизированной фосфоресценции флуорена в н.-гептане, в качестве донора энергии использовался антрон. Спектры сенсибилизированной фосфоресценции флуорена при 77 К не имеют тонкой структуры, однако при понижении температуры до 4,2 К спектры становятся значительно резче.
При исследовании кинетики затухания суммарной интенсивности сенсибилизированной фосфоресценции флуорена в кристаллическом и стеклообразном растворителях в [87] наблюдалось уменьшение времени затухания сенсибилизированной фосфоресценции по сравнению с обычной фосфоресценцией.
В работе [153] исследованы процессы дезактивации колебательно-возбуждённых триплетных молекул флуорена посторонними газами.
Авторами [154] теоретически рассчитаны частоты и интенсивности полос колебаний в спектрах молекул флуорена.
Все используемые соединения марки ”хч” очищались путем двукратной перекристаллизации из растворителя.
2.2 МЕТОДИКА ЭКСПЕРИМЕНТА
Основными экспериментами для решения поставленной задачи являлись изучение кинетики накопления и распада триплетных молекул акцептора и донора энергии, изучение спектров обычной и сенсибилизированной фосфоресценции, а также влияние на них температуры.
Экспериментальная установка была собрана на базе спектрометра ДФС-12 (рис.
3) с дифракционной решёткой 600 штр./мм и линейной дисперсией 5 ![]() /мм.
/мм.
Изучаемый раствор необходимой концентрации помещался в цилиндрическую или лопаточкообразную кварцевую кювету. Исследуемый раствор охлаждался путём быстрого погружения в кварцевый прозрачный сосуд Дьюара с кипящим азотом. Такой способ охлаждения растворов можно назвать быстрым замораживанием в отличие от «медленного» замораживания, которое осуществлялось в парах азота. В последнем случае кристаллизация раствора происходила за 5-10 мин.
Молекулы донора энергии возбуждались светом ртутной лампы ПРК-2 с фильтром 365 нм. Молекулы, используемые в качестве акцепторов энергии, излучение с данной длиной волны не поглощают. При исследовании обычной фосфоресценции молекул акцептора, последние возбуждались излучением ПРК-2 с фильтром 313 нм (аценафтен и флуорен) или 290 нм (нафталин).
Фосфоресценция донора в присутствии молекул акцептора в растворе была сильно потушена. Это позволяло при регистрации спектров сенсибилизированной фосфоресценции обходиться без фосфороскопа.
Градуировка спектрометра производилась по линиям излучения ртутной лампы низкого давления. Ширина входной и выходной щелей монохроматора при записи спектров фосфоресценции была не более 1 мм.

Ошибка в определении максимума 0-0 полосы в спектре сенсибилизированной
фосфоресценции не превышала 5 ![]() .
.
При изучении кинетики сенсибилизированной фосфоресценции для отделения её от фосфоресценции донора использовались электромеханические затворы, управляемые с помощью электронных реле времени. Время срабатывания затворов не превышало 5 мс. Задержка во времени между началом регистрации сенсибилизированной фосфоресценции и прекращением возбуждения донора изменялась от 0,1 до 30 с.
Регистрирующая часть установки включала в себя двухкоординатный графопостроитель Н-307 при записи спектров излучения и кинетики фосфоресценции молекул акцептора. При исследовании кинетики фосфоресценции молекул донора двухкоординатный графопостроитель заменялся на универсальный запоминающий осциллограф С8-13. Для согласования входного сопротивления самописца и выходного сопротивления фотоэлектронного умножителя использовался катодный повторитель, постоянную времени которого можно было изменять от 0,01 до 1,0 с. Линейность работы усилителя постоянного тока проверялась при подаче на ФЭУ светового потока регулируемого изменением входной щели монохроматора. Механическая постоянная времени графопостроителя не превышала 0,03 с.
Кинетические кривые, полученные с помощью графопостроителя или осциллографа перестраивались в полулогарифмическом масштабе, из которого и определялось время разгорания или время затухания фосфоресценции.
Величина погрешности при определении t в экспериментах обуславливалась флуктуациями фототока, нелинейностью усилителя, погрешностью блока временной развёртки и механической постоянной самописца. Три последних источника погрешностей по данным многократных проверок могли дать в сумме систематическую ошибку не более 1,0 %. Для уменьшения влияния флуктуаций фототока измерения повторялись 5-10 раз и общая ошибка в каждом конкретном случае находилась из среднего значения t с учётом возможной систематической ошибки. С учётом вышеизложенного, ошибка при измерении времени затухания сенсибилизированной фосфоресценции не превышала 0,05 с, а времени разгорания – 0,1 с. Большее значение ошибки при измерении времени разгорания обусловлено тем, что флуктуации светового потока источника света влияют на точность измерения времени разгорания и не влияют на точность измерения времени затухания. Ошибка при измерении времени затухания фосфоресценции донора не превышала 0,5 мс.
В температурных исследованиях для уменьшения продольного градиента температуры кварцевая кювета помещалась в медную толстостенную трубочку. При этом для исследования с помощью диафрагмы выделялся участок высотой 2 мм, где находился в образце один из концов дифференциальной термопары. Толщина кюветы была равна 0,3 мм, диаметр – 2 мм. С целью уменьшения влияния поперечного градиента температуры контрольные опыты проводились в лопаточкообразной кювете, толщина исследуемого слоя в которой около 0,5 мм.
Измерение температуры производилось с помощью медь-константановой термопары, проградуированной по точкам плавления н.-парафинов. Один спай термопары находился в сосуде Дьюара с жидким азотом, а второй помещался непосредственно в раствор перед замораживанием. В качестве измерительного прибора использовался гальванометр М-95, с ценой деления 0,01 мВ/дел. Ошибка при измерении температуры не превышала 3 К.
При исследовании температурной зависимости интенсивности и времени затухания сенсибилизированной фосфоресценции нагревание образца происходило в результате испарения азота под образцом. Скорость изменения температуры при этом была около 3 град./мин.
При исследовании температурной зависимости интенсивности сенсибилизированной фосфоресценции спектральная ширина щели бралась максимальной для того, чтобы смещение максимума 0-0 полосы при изменении не превышала её. Это и тот факт, что при увеличении температуры распределение интенсивности в спектре сенсибилизированной фосфоресценции не изменяется, позволяло судить по изменению регистрируемой интенсивности в максимуме 0-0 полосы об изменении интегральной интенсивности.
Отжиг образца производился следующим образом. Полученный в результате быстрого замораживания образец нагревался от 77 К до определённой температуры из области 150-180 К и выдерживался при фиксированной температуре необходимое время (от 0,5 до 40 мин.). Затем образец помещался в жидкий азот, в котором и производилось измерение его люминесцентных характеристик при 77 К.
Для определения влияния отжига на интенсивность сенсибилизированной фосфоресценции записывались спектры фосфоресценции раствора до и после отжига и сравнивались их интегральные интенсивности. Следует заметить, что в этом случае результаты совпадали с результатами, полученными при регистрации сенсибилизированной фосфоресценции в максимуме 0-0 полосы с точностью до 10 %.
При исследовании зависимости интенсивности сенсибилизированной фосфоресценции от времени отжига при фиксированной температуре образец отжигался в течение времени Dt, затем измерялись его люминесцентные характеристики при температуре 77 К. После чего образец снова нагревался до температуры отжига и отжигался в течение времени Dt, в результате чего время его отжига составляло 2Dt. Затем снова измерялись его люминесцентные характеристики. Таким образом, процесс повторялся до тех пор, пока не прекращался рост интенсивности в результате отжига образца.
2.3. ОПРЕДЕЛЕНИЕ ПАРАМЕТРОВ ТРИПЛЕТНОГО СОСТОЯНИЯ МОЛЕКУЛ АКЦЕПТОРА ИЗ КИНЕТИКИ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕЦИИ
Для выяснения механизмов влияния температуры на сенсибилизированную фосфоресценцию необходимо было определить концентрацию триплетных молекул акцептора энергии. В работах М. В. Алфимова с сотрудниками [155,156] была предложена методика определения концентрации триплетных молекул в случае прямого возбуждения органических соединений из кинетических экспериментов:
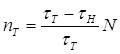 , (13)
, (13)
где tН и tТ – время накопления и распада триплетных молекул, а N – общая концентрация молекул в растворе.
Величина
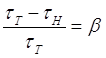 (12)
(12)
показывает, какая часть молекул, участвующая в излучении, находится в триплетном состоянии. В отсутствии реабсорбции излучения величина tН равна времени разгорания фосфоресценции tР, а величина tТ – есть время затухания фосфоресценции. Однако, обоснование возможности применения этой методики в случае сенсибилизированного возбуждения молекул акцептора в литературе отсутствует.
В работах [157,158] предложена методика определения концентрации триплетных молекул органических соединений по изменению интенсивности флуоресценции за счёт уменьшения числа молекул в основном состоянии в результате их перехода в триплетное. Однако эта методика неприменима при сенсибилизированном возбуждении триплетного состояния по причине отсутствия в этом случае флуоресценции.

Рассмотрим кинетику накопления
триплетных молекул акцептора. Для этого используем трехуровневую схему для
молекул донора и двухуровневую для молекул акцептора (рис.4), поскольку
другие энергетические уровни не играют значительной роли в заселении
триплетного уровня молекул акцептора.
Примем следующие обозначения констант скоростей для переходов (в нашем случае константа перехода есть сумма констант излучательного и безызлучательного переходов ):
S0 ® S1 - k0;
S1 ® S0 - k1;
S1 ® T1 - k2;
T1 ® S0 - k3.
Концентрацию молекул обозначим следующим образом: в состоянии S0 - n0, в состоянии S1 - n1, в T1 - n2 (где n2 = nТ).
Скорость изменения концентрации молекул в соответствующем состоянии определяется следующими уравнениями:
![]() ; (15)
; (15)
![]() ; (16)
; (16)
![]() ; (17)
; (17)
![]() ; (18)
; (18)
![]() ; (19)
; (19)
![]() ; (20)
; (20)
![]() , (21),
, (21),
где индексы A
и D указывают на то, что данная величина относится к молекулам
акцептора или донора соответственно; N – общее число молекул в растворе
(на единицу объема), участвующих в данном процессе; kT-T
- константа переноса для пары; k0=IВR (IВ
- интенсивность возбуждающего излучения; R – сечение поглощения); m - величина, показывающая, какая часть
молекул акцептора в основном состоянии ![]() находится
в радиусе обменных взаимодействий с одной триплетной молекулой донора и может
ее потушить в результате переноса энергии; n - величина, показывающая, какая часть триплетных молекул
донора
находится
в радиусе обменных взаимодействий с одной триплетной молекулой донора и может
ее потушить в результате переноса энергии; n - величина, показывающая, какая часть триплетных молекул
донора ![]() находится в радиусе
обменных взаимодействий с одной молекулой акцептора и может ее перевести в
триплетное состояние в результате передачи энергии. Так как число триплетных
молекул донора, перешедших за время dt в основное состояние за счет
переноса энергии равно числу молекул акцептора, перешедших за это время из
основного состояния в триплетное, то
находится в радиусе
обменных взаимодействий с одной молекулой акцептора и может ее перевести в
триплетное состояние в результате передачи энергии. Так как число триплетных
молекул донора, перешедших за время dt в основное состояние за счет
переноса энергии равно числу молекул акцептора, перешедших за это время из
основного состояния в триплетное, то
![]() , (22)
, (22)
а следовательно
m =n. (23)
Система дифференциальных уравнений (15)-(21) является нелинейной, и поэтому найти ее решение в общем случае сложно. Однако можно найти приближённое решение для частного случая, который часто реализуется в экспериментальных исследованиях.
Действительно, для удобства экспериментальных исследований кинетики и
спектров сенсибилизированной фосфоресценции молекул в замороженных растворах
обычно берут донорно-акцепторные пары, которые удовлетворяют следующему
условию: константа перехода ![]() для
молекул донора на несколько порядков больше соответствующей константы
для
молекул донора на несколько порядков больше соответствующей константы ![]() для молекул акцептора
[7,87]:
для молекул акцептора
[7,87]:
![]() »
» ![]() . (24)
. (24)
Как отмечалось в 2.1, это позволяет разделить во времени фосфоресценцию акцептора и донора.
Предварительные экспериментальные исследования кинетики разгорания сенсибилизированной фосфоресценции, а также результаты работы [87] показывают, что для таких систем время разгорания сенсибилизированной фосфоресценции соизмеримо со временем жизни триплетных молекул акцептора. Следовательно, для таких пар выполняется неравенство
![]() «
« ![]() . (25)
. (25)
При выполнении условия (25) в первом приближении можно пренебречь
дезактивацией энергии триплетного возбуждения в молекулах донора за счет
передачи энергии акцептору при рассмотрении кинетики их накопления. Тогда в
уравнениях (15) и (17) последние члены можно отбросить и система уравнений (15)
– (17) становится линейной. Одновременное выполнение наряду с (25) условия (24)
позволяет считать в уравнениях (19) и (20) величину ![]() постоянной,
равной
постоянной,
равной
![]() , (26)
, (26)
поскольку динамическое равновесие заселенности состояний в молекулах донора устанавливается за время намного меньшее, чем в молекулах акцептора. Поэтому константу перехода молекул акцептора из основного состояния в триплетное можно обозначить
![]() (27)
(27)
и считать величиной постоянной.
Таким образом, при выполнении условий (24) и (25) систему уравнений (15) – (21) можно представить как две системы линейных уравнений:
![]()
![]() ; (15а)
; (15а)
![]() ; (16а)
; (16а)
![]() ; (17а)
; (17а)
![]() ; (18а)
; (18а)
![]()
![]() ; (19а)
; (19а)
![]() ; (20а)
; (20а)
![]() . (21а)
. (21а)
Система уравнений (15а) – (18а) описывает динамику распределения молекул донора, а система (19а) – (21а) – динамику распределения молекул акцептора по энергетическим уровням.
Решение системы (19а) – (21а) с учетом (26) будет иметь вид
![]() . (28)
. (28)
Из (28) получаем значение стационарной заселенности (при t ® ¥)
![]() . (29)
. (29)
В отсутствие реабсорбции излучения и других каналов дезактивации энергии
триплетного возбуждения в молекулах акцептора интенсивность сенсибилизированной
фосфоресценции I(t)
пропорциональна ![]() , поэтому можно
записать для сенсибилизированной фосфоресценции
, поэтому можно
записать для сенсибилизированной фосфоресценции
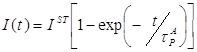 , (30)
, (30)
где IST-
максимальное значение интенсивности сенсибилизированной фосфоресценции( при t® ¥),
![]() -время разгорания, равное
-время разгорания, равное
![]() . (31)
. (31)
На рис. (5) приведена экспериментальная кривая разгорания
сенсибилизированной фосфоресценции аценафтена в н.-гексане при 77 К. Как видно,
экспериментальные точки хорошо укладываются на теоретическую кривую,
описываемую уравнением (30) с ![]() =1,16 с.
=1,16 с.

Зависимость интенсивности сенсибилизированной фосфоресценции (стационарной) от мощности возбуждения, как следует из (29) и (27), можно представить в виде
![]() , (32)
, (32)
где ![]() - постоянные величины. В
выражении (32) от мощности возбуждения зависит величина
- постоянные величины. В
выражении (32) от мощности возбуждения зависит величина![]() .
.
Решение системы уравнений (15а)-(18а) дает зависимость ![]() от возбуждающего света
от возбуждающего света
![]() . (33)
. (33)
Введя соответствующие обозначения: ![]() ,
,
![]()
![]() и подставляя (33) в (32),
окончательно получим
и подставляя (33) в (32),
окончательно получим
![]() . (34)
. (34)
Таким образом, формально зависимость интенсивности сенсибилизированной фосфоресценции от мощности возбуждения (34) совпадает с зависимостью интенсивности обычной фосфоресценции от интенсивности возбуждения [159, 160].
Рассмотрим кинетику дезактивации триплетных молекул акцептора при выполнении условий (24) и (25). В этом случае дезактивацией триплетного состояния молекул донора за счет переноса энергии на акцептор можно пренебречь, поэтому изменение концентрации триплетных молекул акцептора со временем будет происходить по закону:
![]() . (35)
. (35)
Решением этого уравнения с учетом (29) является
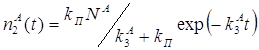 . (36)
. (36)
Соответственно закон затухания сенсибилизированной фосфоресценции для донорно-акцепторных пар, удовлетворяющих условиям (24) и (25) носит экспоненциальный характер:
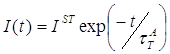 , (37)
, (37)
где
![]() (38)
(38)
- время затухания сенсибилизированной фосфоресценции акцептора.
Подставляя (31) и (38) в (29), имеем:
![]() (39)
(39)
и для стационарной концентрации триплетных молекул акцептора можем записать:
![]() . (40)
. (40)
Полученное равенство позволяет найти стационарную концентрацию триплетных молекул акцептора энергии из кинетических экспериментов.
Для проверки полученных теоретических выводов было проведено экспериментальное исследование зависимости концентрации триплетных молекул аценафтена (акцептор энергии) от интенсивности возбуждающего света. Интенсивность возбуждающего света изменялась с помощью нейтральных фильтров (калиброванных металлических сеток), а концентрация триплетных молекул определялась по формуле (40). Экспериментальные результаты приведены на рис.6, где по оси абсцисс отложена величина, обратная интенсивности возбуждающего света IВ. За единицу приято максимальное возбуждение, которое соответствует относительной концентрации триплетных молекул 0,5. По оси ординат отложена величина, обратная относительной заселённости триплетного уровня молекул акцептора.
Как видно из рисунка, экспериментальные точки хорошо укладываются на прямую, что согласуется с выражением (34).
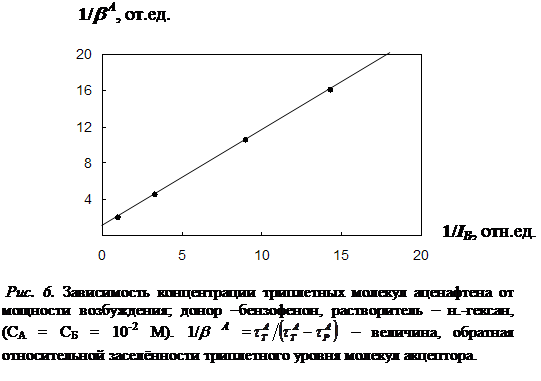 |
Полученное равенство (40) не только позволяет найти концентрацию триплетных молекул акцептора из кинетических параметров, но и определить константу перехода молекул акцептора в триплетное состояние
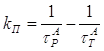 . (41)
. (41)
На основании всего вышесказанного можно сделать следующие выводы.
Для
донорно-акцептроных пар, удовлетворяющих условиям (24)–(26), закон разгорания
сенсибилизированной фосфоресценции имеет экспоненциальный характер. Определенные
из кинетических экспериментов параметры – время разгорания и затухания сенсибилизированной
фосфоресценции – позволяют определить по формуле (40) долю молекул акцептора в
триплетном состоянии от общего числа, участвующих в триплет-триплетном
переносе энергии (![]() ).
).
Формула (40) для определения концентрации триплетных молекул акцептора совпадает с формулой, полученной ранее М.В.Алфимовым с сотрудниками, для определения концентрации молекул в триплетном состоянии при возбуждении быстрыми электронами [156]. Однако время разгорания сенсибилизированной фосфоресценции зависит не только от интенсивности возбуждения, но и от константы переноса энергии в донорно-акцепторной паре.
Зависимость интенсивности сенсибилизированной фосфоресценции от мощности возбуждения в указанном выше приближении определяется выражением (34), которое по форме совпадает с зависимостью интенсивности фосфоресценции от мощности возбуждения при фотовозбуждении.
Выражение (41) позволяет вычислить константу перехода молекул акцептора из основного состояния в триплетное в результате переноса энергии.
Глава 3
Особенности температурной зависимости параметров сенсибилизированной фосфоресценции примесных молекул в замороженных н.-парафиновых растворах
В данной главе приведены результаты исследования температурной зависимости параметров сенсибилизированной фосфоресценции ароматических углеводородов в замороженных н.-парафиновых растворах. Показано, что при повышении температуры изменение интенсивности сенсибилизированной фосфоресценции носит немонотонный характер, включая как интервалы уменьшения, так и увеличения. Здесь же приведены результаты исследования влияния концентрации примесей и растворителя на характер температурной зависимости сенсибилизированной фосфоресценции для различных донорно-акцепторных пар. На основании этих результатов сделан вывод об общности немонотонного характера температурной зависимости интенсивности сенсибилизированной фосфоресценции ароматических углеводородов в замороженных н.-парафиновых растворах. Установлено, что увеличение интенсивности сенсибилизированной фосфоресценции в результате нагревания раствора является необратимым. Выдвинуто предположение о наличии процесса, приводящего к снятию концентрационного тушения сенсибилизированной фосфоресценции в результате нагревания, скорость которого зависит от температуры.
3.1 ТЕМПЕРАТУРНАЯ ЗАВИСИМОСТЬ ИНТЕНСИВНОСТИ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ ОРГАНИЧЕСКИХ МОЛЕКУЛ
Интенсивность фосфоресценции пропорциональна константе излучательного перехода из нижнего триплетного состояния в основное синглетное состояние и концентрации триплетных молекул. Если вероятность излучательного перехода не изменяется, то характер изменения интенсивности фосфоресценции и характер изменения концентрации триплетных молекул совпадают.
Для исследуемых систем изменения констант излучательных переходов при изменении температуры не наблюдалось [7], поэтому, в дальнейшем будем считать, что в отсутствие реабсорбции излучения, характер изменения интенсивности сенсибилизированной фосфоресценции обусловлен изменением концентрации триплетных молекул акцептора энергии.
В общем плане температурных исследований прежде всего необходимо было установить характер зависимости интенсивности сенсибилизированной фосфоресценции от температуры. При сенсибилизированном заселении триплетного уровня молекул акцептора могут существовать дополнительные каналы дезактивации возбуждения к тем каналам, которые имеются при прямом возбуждении в отсутствие переноса энергии. Если скорость деградации энергии триплетного возбуждения через дополнительные каналы зависит от температуры, то их наличие прежде всего можно обнаружить, исследуя зависимость интенсивности сенсибилизированной фосфоресценции от температуры. С этой целью в первую очередь проведено сравнение температурной зависимости интенсивности и длительности сенсибилизированной фосфоресценции молекул акцептора и фосфоресценции молекул донора в отсутствии и в присутствии молекул акцептора.
Обнаружено, что характер температурной зависимости интенсивности сенсибилизированной фосфоресценции существенно отличается от обычной фосфоресценции.
Температурная зависимость интенсивности I и длительности t сенсибилизированной фосфоресценции аценафтена в н.-гексане, когда донором являлся бензофенон, по отношению к значению этих величин I0 и t0 при 77 К соответственно представлены на рис.7. Ниже указаны используемые концентрации бензофенона - СБ и аценафтена – СА в растворе. Как видно из рисунка, зависимость интенсивности сенсибилизированной фосфоресценции от температуры имеет немонотонный характер.
В области от 77 до 155 К (область 1) сенсибилизированная фосфоресценция испытывает температурное тушение. Уменьшение интенсивности I сопровождается незначительным падением t . Более медленное изменение t в сравнении с I , по-видимому, обусловлено тем, что величина I уменьшается за счёт тушения триплетных молекул как акцептора, так и донора энергии. Изме
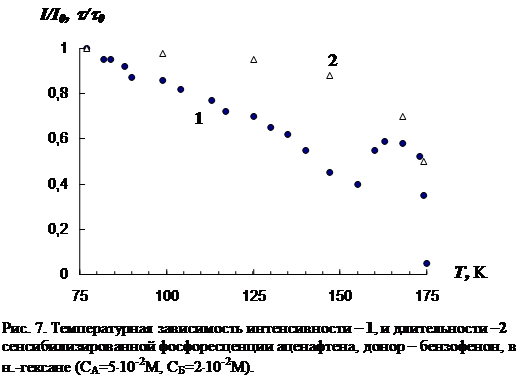 |
нение же величины t происходит только за счёт тушения триплетных состояний молекул акцептора. В случае обычной фосфоресценции при повышении температуры интенсивность фосфоресценции также уменьшается, и как было показано в работах Гребенщикова Д. М. и Персонова Р. И. [25,119], основной вклад в температурное тушение вносит диффузионное кислородное тушение, хотя не исключены и другие механизмы.
Особенностью для области 155-167 К (область 2) является рост интенсивности I в процессе нагревания образца, сопровождающийся уменьшением t . Падение t указывает на то, что процесс тушения продолжается и в этой области, однако рост интенсивности свидетельствует о начале какого-то процесса, ведущего к увеличению числа триплетных молекул акцептора.
Дальнейшее эффективное тушение связано с плавлением н.-гексана (178К).
Увеличение интенсивности в температурной области 2 является нехарактерным для обычной фосфоресценции исследуемых молекул в н.-парафиновых растворах [25,117-120]. Интенсивность обычной фосфоресценции при повышении температуры либо не изменяется, либо уменьшается.
Для того, чтобы установить, как влияет присутствие акцептора в растворе на характер температурной зависимости фосфоресценции донора, был проведен соответствующий эксперимент для той же пары бензофенон-аценафтен в н.-гексане.
На рис. 8 представлена температурная зависимость относительной интенсивности фосфоресценции молекул донора (I/I0, где I0 – интенсивность при 77 К) в отсутствие молекул акцептора (кривая 1) и в присутствие при различных концентрациях акцептора (кривые 2 и 3).
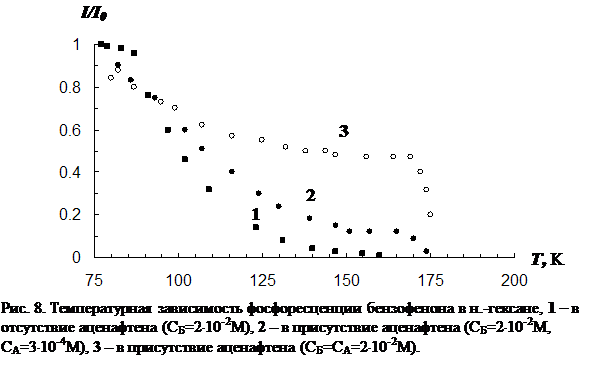 |
Как видно из рисунка, характер температурной зависимости фосфоресценции донора во всех случаях не повторяет наблюдаемого для сенсибилизированной фосфоресценции. Во всём исследуемом диапазоне температур от 77 до 178 К, фосфоресценция донора испытывает тушение.
Фосфоресценция бензофенона в отсутствие аценафтена испытывает более эффективное тушение, чем в его присутствие. Характер тушения фосфоресценции бензофенона в отсутствие молекул аценафтена хорошо совпадает с результатами кислородного тушения фосфоресценции примесных молекул, вытесненных на поверхность кристалликов [25,117-120]. Резкое уменьшение I/I0 (кривая 1) начинается при Т » 90 К (точка кипения кислорода) и тушится практически полностью уже при 150 К.
В присутствии молекул акцептора в растворе характер тушения фосфоресценции донора существенно отличается от кислородного. Увеличение концентрации акцептора в растворе ведёт к тому, что тушение становится менее эффективным (кривая 3). Однозначного объяснения такому поведению интенсивности фосфоресценции донора дать сложно. Предположительно, это можно объяснить тем, что присутствие молекул акцептора препятствует взаимодействию молекул донора с кислородом, чем уменьшает диффузионное кислородное тушение. С другой стороны, обращает на себя внимание заметное отличие характера тушения в области температур 150-175 К (кривые 2 и 3). Можно предположить, что при температуре выше 150 К существует процесс, который компенсирует кислородное тушение, т.е. наряду с кислородным тушением существуют механизмы, приводящие к увеличению интенсивности фосфоресценции донора.
Спектр обычной фосфоресценции аценафтена в н.-гексане при 77 К является квазилинейчатым, за который ответственны молекулы акцептора, внедрённые в кристаллики растворителя. Молекулы аценафтена, на которые происходит эффективный перенос энергии, находятся на поверхности кристалликов или в пространстве между ними. Поэтому большая интенсивность квазилинейчатого спектра не позволяет исследовать температурную зависимость обычной фосфоресценции аценафтена в отсутствие молекул бензофенона в растворе.
Таким образом, характер температурной зависимости фосфоресценции молекул бензофенона в присутствие молекул аценафтена в растворе и сенсибилизированной фосфоресценции молекул аценафтена в н.-гексане отличаются от характера температурной зависимости бензофенона в отсутствие молекул акцептора. Увеличение интенсивности сенсибилизированной фосфоресценции при повышении температуры нехарактерно для обычной фосфоресценции примесных молекул в н.-парафиновых растворах и поэтому температурная область 2 была названа аномальной.
С целью ответа на вопрос – насколько общим является немонотонный характер температурной зависимости сенсибилизированной фосфоресценции, была исследована эта зависимость для других донорно-акцепторных пар в данном растворителе.
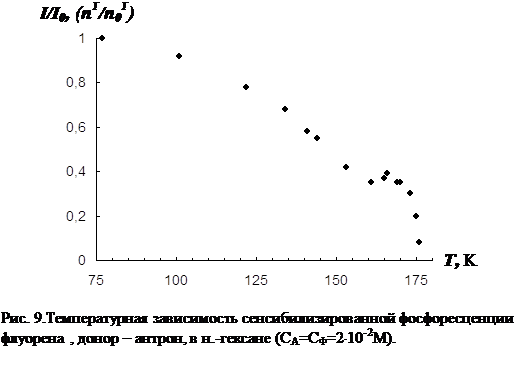 |
На рис.9 представлена температурная зависимость относительной интенсивности сенсибилизированной фосфоресценции I/I0 (что совпадает с изменением относительной концентрации триплетных молекул nT/n0T, где n0Т – концентрация триплетных молекул акцептора при 77 К) флуорена в н.-гексане, когда донором являлся антрон. Качественно характер кривой совпадает с таковым для донорно-акцепторной пары аценафтен-бензофенон (рис.7).
При изменении температуры от 77 до 160 К интенсивность сенсибилизированной фосфоресценции уменьшается. При повышении температуры от 160 К до 166 К наблюдается незначительное увеличение I/I0 (nT/n0T). Начиная со 166 К и до точки плавления растворителя наблюдается эффективное тушение фосфоресценции молекул акцептора.
На рис.10 (кривая 1) приведена зависимость I/I0 (nT/n0T) для пары бензофенон-нафталин в н.-гексане. Как видно из рисунка, в этом случае температурная зависимость интенсивности сенсибилизированной фосфоресценции имеет так же немонотонный характер. Увеличение I/I0 (nT/n0T) наблюдается в интервале температур от 158 до 170 К.
На основании этих результатов был сделан вывод об общем характере такой температурной зависимости интенсивности сенсибилизированной фосфоресценции ароматических углеводородов в н.-гексане.
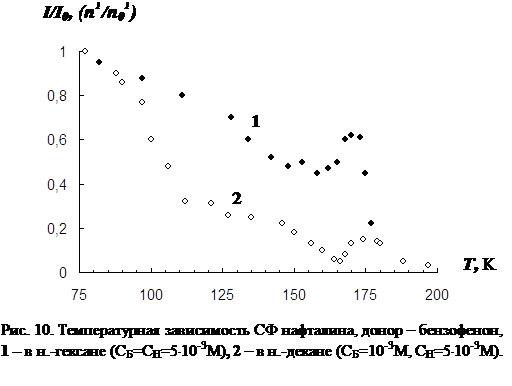 |
Область увеличения интенсивности сенсибилизированной фосфоресценции для всех исследованных донорно-акцепторных пар в н.-гексане лежит вблизи точки плавления растворителя. Необходимо было проверить предположение, не является ли увеличение интенсивности сенсибилизированной фосфоресценции в температурной области 2 следствием каких-либо динамических процессов, предшествующих плавлению растворителя. Для проверки данной гипотезы была исследована температурная зависимость интенсивности сенсибилизированной фосфоресценции примесных молекул в н.-октане и н.-декане, точки плавления которых лежат намного выше, чем н.-гексана (табл. 1).
На рис.10 (кривая 2) изображена температурная зависимость I/I0 сенсибилизированной фосфоресценции нафталина в н.-декане, донор – бензофенон. Температурная область 1, где наблюдается тушение I/I0, в данном опыте лежит в более широком диапазоне – 77- 165 К. При достижении конечной температуры из этой области наблюдается почти полное тушение сенсибилизированной фосфоресценции, I/I0 падает до значения 0.05. Такое сильное тушение не было обнаружено ни для одной донорно-акцепторной пары в н.-гексане (рис. 7, 9, 10 (кривая 1)), где величина I/I0 в конце температурной области 1 достигала значений от 0.35 до 0.45. При этом в н.-гексане использовались более высокие концентрации примесей, чем в н.-декане. Температурная область 2, в которой происходит рост интенсивности, 165 –175 К, в н.-декане лежит намного ниже точки плавления растворителя –243 К. От 175 К происходит дальнейшее уменьшение I/I0, уже при 188 К достигающее значение 0.05.
На рис.11 приведена температурная зависимость I/I0 (nT/n0T) аценафтена в н.-октане (кривая 2) и в н.-гексане (кривая 1) с целью их сравнения, когда донором являлся бензофенон. Концентрация примесей в н.-октане для данного опыта была выбрана меньшей (СБ=СА= 2.5×10-3 М), где СБ и СА – концентрации донора и акцептора соответственно), чем в н.-гексане (СБ=СА=10-2 М).
Диапазон температурной области 1 в обоих растворителях с точностью до ошибки опыта (±3 К) совпадает, 77 – 150 К. В н.-октане значение I/I0 в конце области 1 падает до 0.07, в н.-гексане – до 0.46. Начало роста концентрации триплетных молекул в н.-октане совпадает с н.-гексаном, максимум так же достигается при одинаковой температуре – 171 К. Однако величина максимума I/I0 в н.-октане больше – 1.51, чем в н.-гексане – 0.46 (при этом следует отметить, что концентрация примесей в н.-гексане больше, чем в н.-октане). Аномальная температурная область в н.-октане лежит намного ниже точки плавления растворителя – 216 К. Более быстрое тушение сенсибилизированной фосфоресценции в н.-гексане, чем в н.-октане связано с более низкой точкой плавления н.-гексана в сравнении с н.-октаном.
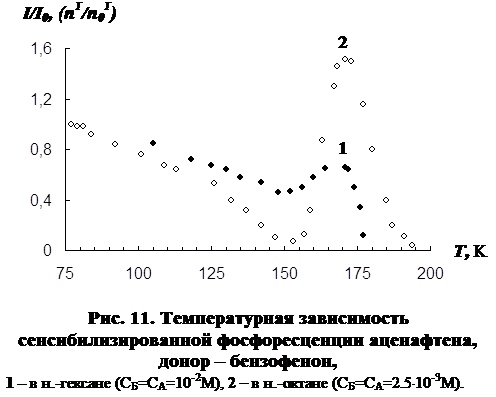 |
Таким образом, температурный интервал, в котором наблюдается увеличение интенсивности сенсибилизированной фосфоресценции при повышении температуры не зависит от точки плавления растворителя. На основании этого можно сделать вывод, что такой характер температурной зависимости интенсивности сенсибилизированной фосфоресценции не обусловлен процессами, предшествующими плавлению растворителя.
Аномальный характер температурной зависимости сенсибилизированной фосфоресценции наблюдался для всех изученных донорно-акцепторных пар во всех используемых растворителях. Поэтому можно говорить об общности немонотонного характера температурной зависимости сенсибилизированной фосфоресценции ароматических углеводородов в замороженных н.-парафиновых растворах.
Из представленных экспериментальных данных видно, что изменение величины I/I0 (nT/n0T) в температурной области 2 для одной и той же донорно-акцепторной пары зависит как от концентрации примесей, так и от растворителя (см. рис 10,11). Поэтому с целью выяснения причин аномального характера температурной зависимости сенсибилизированной фосфоресценции необходимо было более подробно исследовать влияние концентрации донорно-акцепторной смеси и растворителя на неё.
3.2 ВЛИЯНИЕ КОНЦЕНТРАЦИИ ПРИМЕСЕЙ И РАСТВОРИТЕЛЯ НА ПАРАМЕТРЫ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ
При исследованиях влияния концентрации донорно-акцепторной смеси на характер температурной зависимости сенсибилизированной фосфоресценции были выбраны пары бензофенон-нафталин в н.-гексане (рис.12) и бензофенон-аценафтен в н.-октане (рис.13).
Для пары бензофенон-нафталин в н.-гексане концентрация молекул донора в растворе равнялась концентрации молекул акцептора и изменялась от 5×10-3 М (рис.12, кривая 1 ) до 10-2 М (рис.12, кривая 2). Как видно из рисунка, увеличение интенсивности сенсибилизированной фосфоресценции в аномальной области 2 при повышении температуры тем больше, чем выше концентрация молекул примесей в растворе. Чтобы проверить, насколько общим является такое влияние концентрации на увеличение интенсивности сенсибилизированной фосфоресценции в аномальной температурной области, была исследована температурная зависимость для другой пары в другом растворителе при различных концентрациях раствора.
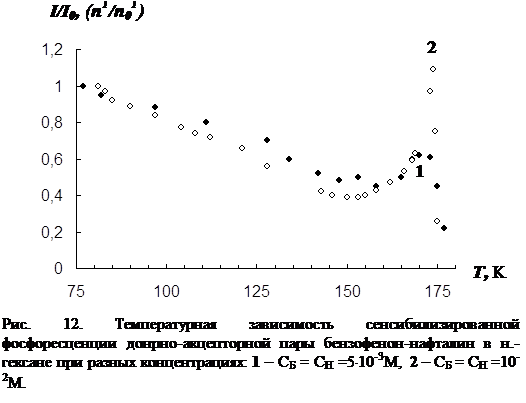 |
На рис.13 представлена температурная зависимость сенсибилизированной фосфоресценции аценафтена в н.-октане для трёх различных его концентраций при одной и той же концентрации бензофенона. В этом случае, как и в предыдущем для нафталина, наблюдается большее увеличение интенсивности сенсибилизированной фосфоресценции аценафтена в аномальной температурной области для больших концентраций.
Таким образом, можно сделать вывод, что при повышении концентрации молекул примеси в растворе эффект увеличения интенсивности сенсибилизированной фосфоресценции примесных молекул в н.-парафиновых растворах при нагревании в температурной области 2 усиливается.
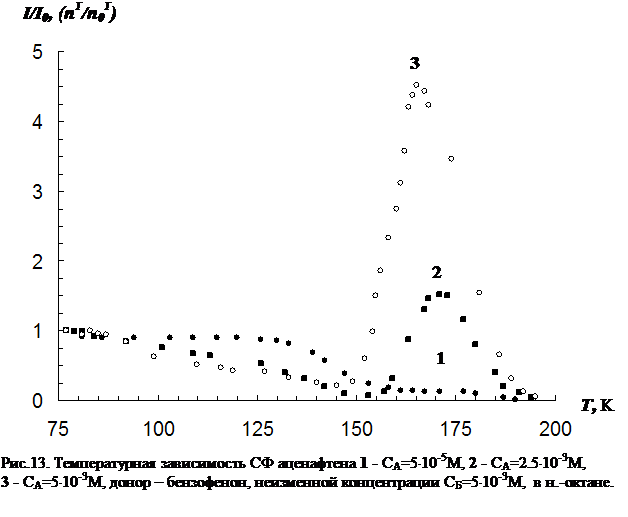 |
Как уже отмечалось в 3.1, по-видимому, в замороженных н.-парафиновых растворах донорно-акцепторных смесей существует два процесса, влияющих на концентрацию триплетных молекул акцептора энергии, а следовательно и на интенсивность сенсибилизированной фосфоресценции.
Первый процесс – тушение триплетных состояний молекул донора акцептором энергии, приводит к падению интенсивности сенсибилизированной фосфоресценции при повышении температуры и сопровождается падением t . Падение интенсивности сенсибилизированной фосфоресценции в результате этого процесса происходит во всём исследованном интервале температур, в том числе и в аномальной области 2. Этот процесс практически не зависит от концентрации молекул примеси в растворе.
Второй процесс ведет к увеличению числа триплетных молекул. Прирост числа триплетных молекул акцептора энергии в результате этого процесса зависит как от концентрации раствора (рис.12, 13), так и от температуры.
При определенных (высоких) концентрациях молекул примеси в растворе интенсивность сенсибилизированной фосфоресценции в результате второго процесса увеличивается в большее число раз, чем она уменьшается в результате тушения.
Наличием таких двух процессов можно объяснить и другие, менее заметные концентрационные изменения в температурных зависимостях интенсивности сенсибилизированной фосфоресценции, представленные на рис. 12 и 13.
Так, увеличение интенсивности сенсибилизированной фосфоресценции при повышении температуры становится заметным тем раньше, чем выше концентрация раствора. Для пары бензофенон-нафталин увеличение интенсивности сенсибилизированной фосфоресценции начинается при 150 К для концентраций СБ = СН = 10-2 М и при 158 К для концентрации СБ = СН = 5×10-3 М (рис.12).
При одних и тех же концентрациях молекул примеси в растворе для пары бензофенон-аценафтен в н.-октане СБ = СН = 5×10-3 М (рис.13) и пары бензофенон-нафталин в н.-гексане (рис.12) увеличение интенсивности сенсибилизированной фосфоресценции начинается раньше для первой пары (при Т=145 К). Для этой же пары максимальное увеличение интенсивности сенсибилизированной фосфоресценции в температурной области 2 значительно больше, чем для второй.
Аномальная температурная зависимость сенсибилизированной фосфоресценции наблюдается не для всех концентраций примесей. При значительном уменьшении концентрации акцептора ход температурной кривой сглаживается, область увеличения интенсивности отсутствует (рис. 13, кривая 1).
В связи с этим, возникла необходимость изучения люминесцентных характеристик донорно-акцепторных смесей при различных их концентрациях.
Для пары бензофенон-аценафтен в н.-гексане измерялись интенсивность фосфоресценции бензофенона (рис. 14, кривая 1) и интенсивность сенсибилизированной фосфоресценции аценафтена (рис. 14, кривая 2) по отношению к I0Б (интенсивность фосфоресценции бензофенона при концентрации аценафтена 10-4 М) при изменении концентрации акцептора от 10-4 до 10-2 М. Концентрация бензофенона в растворе была 10-2 моль/л и не изменялась в процессе опыта. Измерения производились при 77 К.
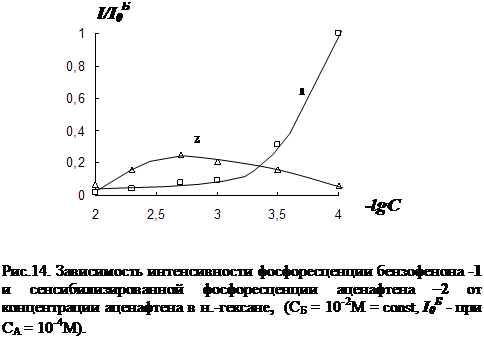 |
Как видно из рисунка, с увеличением концентрации молекул аценафтена в растворе интенсивность фосфоресценции бензофенона уменьшается во всем исследованном интервале концентраций (кривая 1). Такая зависимость качественно объясняется увеличением числа молекул донора энергии попадающих в сферу тушения молекул акцептора при повышении концентрации последних [7].
По характеру концентрационной зависимости относительной интенсивности сенсибилизированной фосфоресценции аценафтена кривую 2 можно разбить на 2 области. Первая область соответствует концентрации аценафтена в растворе от 10-4 моль/л до 5×10-3 моль/л. Вторая область от 5×10-3 моль/л до 10-2 моль/л.
В первой области интенсивность сенсибилизированной фосфоресценции растет с увеличением концентрации молекул аценафтена в растворе. Рост интенсивности сенсибилизированной фосфоресценции в этой области сопровождается падением интенсивности свечения донора энергии. Из анализа кривых 1 и 2 следует согласно [7], что квантовый выход сенсибилизированной фосфоресценции в этой области в пределах точности эксперимента не зависит от концентрации молекул акцептора и начинает изменяться лишь вблизи её значений 5×10-3 моль/л. Этот результат хорошо согласуется с выводами теории переноса энергии триплетного возбуждения между отдельными молекулами примеси, в отсутствие их агрегации [7,21].
Во второй области интенсивность сенсибилизированной фосфоресценции молекул аценафтена резко падает с увеличением их концентрации в растворе, т.е. наблюдается её концентрационное тушение. При наличии концентрационного тушения квантовый выход сенсибилизированной фосфоресценции также уменьшается с повышением концентрации молекул акцептора в растворе.
Сравнение экспериментальных результатов показывает, что аномальная температурная зависимость и концентрационное тушение сенсибилизированной фосфоресценции наблюдается при одинаковых концентрациях акцептора в растворе.
Наряду с концентрационным изменением интенсивности сенсибилизированной фосфоресценции было исследовано поведение других люминесцентных характеристик – положение максимума 0-0 полосы и времени затухания сенсибилизированной фосфоресценции при изменении концентрации примесей в растворе.
В таблице 4 приведена концентрационная зависимость положения максимума 0-0 полосы (lmax) и времени затухания (tТ) сенсибилизированной фосфоресценции пары бензофенон-аценафтен в н.-гексане.
Таблица 4
Концентрационная зависимость параметров сенсибилизированной фосфоресценции аценафтена, донор – бензофенон, в н.-гексане.
|
СБ=СА, М |
10-2 |
5×10-3 |
2.5×10-3 |
10-4 |
5×10-4 |
2.5×10-3 |
10-4 |
|
lmax, нм |
481.2 | 481.0 | 480.7 | 480.6 | 480.4 | 480.2 | 479.6 |
|
tТ, с |
2.25 | 2.30 | 2.40 | 2.50 | 2.55 | 2.65 | 2.65 |
Из данных таблицы видно, что максимум 0-0 полосы при увеличении концентрации молекул примеси смещается в длинноволновую область, время затухания при этом уменьшается.
В ходе проведённых экспериментов было замечено, что интервал концентраций молекул примесей, в котором наблюдается аномальная температурная зависимость интенсивности сенсибилизированной фосфоресценции, отличается для различных растворителей.
В н.-гексане для получения аномальной температурной зависимости I/I0 необходимо создавать довольно высокие концентрации молекул примесей. При этом в температурной области 2 наблюдается лишь незначительное увеличение относительной интенсивности, не превышающее первоначальное. Так, например, для донорно-акцепторной пары бензофенон-аценафтен при концентрациях бензофенона и аценафтена - 10-2 М (рис.11, кривая 1) наблюдается увеличение I/I0 в температурной области 2 от 0.47 до 0.66; при концентрации бензофенона - 2×10-2 и аценафтена - 5×10-2 (рис.7) наблюдается увеличение I/I0 от 0.4 до 0.6. Достичь большего увеличения I/I0 за счёт увеличения концентрации примесных молекул не удаётся из-за образования при их повышении в растворе других центров, отличных от мономерных молекул.
В н.-октане диапазон концентраций примесей, при которых наблюдается аномальный температурный ход кривой, сдвинут в сторону меньших концентраций. Для пары бензофенон-аценафтен уже при концентрации примесей 2.5×10-3 М в н.-октане наблюдается увеличение I/I0 в температурной области 2 от 0.07 до 1.50 (рис.11, кривая 2). При увеличении концентраций донора и акцептора до 5×10-3 М наблюдается изменение I/I0 от 0.2 до 4.7 (рис. 13, кривая 3).
Можно сделать вывод, что для донорно-акцепторной пары бензофенон-аценафтен переход от н.-гексана к н.-октану ведёт к более выраженной аномальной температурной зависимости I/I0.
Рассматривая причины, которые могут обуславливать такое влияние растворителя, представляется важным рассмотреть вопрос о расположении молекул примеси при замораживании в н.-парафиновых матрицах. Такую возможность даёт анализ структуры спектров молекул примесей в каждом из растворителей.
В таблице 5 собраны данные по исследованию структуры спектров люминесценции [86,115,148] выбранных соединений в растворителях от н.-гексана до н.-декана при низких температурах.
Таблица 5.
Виды спектров ароматических углеводородов в н.-парафиновых матрицах при низких температурах.
СоединениеРастворитель |
||||
| н.-гексан | н.-гептан | н.-октан | н.-декан | |
| Бензофенон |
Д[86] |
Д[86] |
Д | Д |
| Антрон |
К[86] |
К[86] |
- | - |
| Аценафтен |
К[148] |
К[148] (10-2-10-3М) Д[148] (10-4-10-5М) |
Д[148] |
Д |
| Нафталин |
К[115] (10-2М) Д[115] (10-3-10-5М) |
Д[115] |
Д[115] |
Д |
| Флуорен |
К[86] |
К[86] |
- | - |
П р и м е ч а н и е. Д – диффузионный спектр, К – квазилинейчатый спектр;
[85], [115], [148] – ссылки на источник литературы; без ссылок – данные настоящей работы.
Из таблицы видно, что для антрона и флуорена н.-гексан и н.-гептан являются «удобными» растворителями.
Для бензофенона, который использовался в качестве донора энергии, все используемые растворители являются «неудобными», т.е. в процессе кристаллизации практически все молекулы бензофенона вытесняются на поверхность.
Аценафтен и нафталин, используемые как акцепторы, в исследуемых растворителях обнаруживают различный характер спектров. В н.-гексане аценафтен при любых концентрациях от 10-5 до 10-2 М имеет квазилинейчатый спектр. В н.-гептане вид спектра зависит от концентрации аценафтена: в диапазоне 10-3 - 10-2 М – спектр квазилинейчатый, 10-4 – 10-5 М – спектр диффузный. Причины такой зависимости обсуждались выше. В н.-октане и н.-декане спектры аценафтена при любых концентрациях имеют диффузный характер [148].
Нафталин уже в н.-гексане обнаруживает зависимость вида спектра от концентрации: при 10-2 М – спектр квазилинейчатый, в диапазоне 10-3 – 10-5 М –диффузный. Далее, от н.-гептана до н.-декана растворители являются «неудобными» для нафталина [115].
Анализируя приведённые данные по структуре спектров, можно обнаружить связь между «удобством» растворителя, для каждого из входящих в донорно-акцепторную пару соединений, и величиной увеличения числа триплетных молекул акцептора в аномальной температурной области.
Получить аномальную температурную зависимость интенсивности сенсибилизированной фосфоресценции для пары антрон-флуорен в н.-гексане удалось только для больших концентраций - 2×10-2 М (рис.9). Причём увеличение составляло всего лишь несколько процентов от первоначального значения. Такая концентрация примеси является одной из максимальных, а значение увеличения интенсивности – минимальным для всех исследованных донорно-акцепторных пар. При этом и для донора и для акцептора энергии данный растворитель является «удобным».
Для пар бензофенон-аценафтен и бензофенон-нафталин в н.-гексане и н.-гептане наблюдается немного большее увеличение интенсивности, чем для пары антрон-флуорен, при меньших концентрациях. Так, например интенсивность сенсибилизированной фосфоресценции аценафтена в н.-гексане в аномальной температурной области увеличивается на 20%, при этом концентрация молекул примесей составляла 10-2 М (рис.11, кривая 1). В данной паре н.-гексан является «неудобным» растворителем для донора и «удобным» для акцептора. Сенсибилизированная фосфоресценция нафталина в н.-гексане при такой же концентрации примесных молекул, 10-2 М, увеличивается в аномальной температурной области до значения, превышающее первоначальное при 77 К (рис.12, кривая 1). Условия «удобства» н.-гексана для нафталина более сложные, чем для аценафтена, т.е. предельная концентрация молекул нафталина, до которой наблюдается внедрение молекул примеси в матрицу растворителя, меньше. Таким образом, «неудобные» растворители способствуют увеличению максимума температурной кривой I/I0.
Н.-октан и н.-декан для донора – бензофенона и акцепторов – аценафтена и нафталина являются «неудобными» растворителями. Наряду с этим в данных растворителях наблюдается максимальное увеличение числа триплетных молекул в температурных измерениях при наименьших исследуемых концентрациях 10-3 - 5×10-3 М.
При высоких концентрациях в «удобных» растворителях часть молекул внедряется в кристаллы растворителя, а часть вытесняется. В неудобных же растворителях практически все молекулы вытесняются на поверхность. Поэтому локальная концентрация молекул примеси на поверхности кристаллов будет больше в неудобных растворителях, чем в удобных при одной и той же средней концентрации. Этим, по-видимому, объясняется то, что в неудобных растворителях аномальная температурная зависимость наблюдается при меньших концентрациях и более чётко выражена. Это даёт основание предположить, что процесс, ответственный за увеличение числа триплетных молекул, происходит на поверхности кристаллов. А так как сенсибилизированная фосфоресценция наблюдается в результате переноса энергии, то молекулы донора и акцептора, участвующие в процессе переноса находятся на поверхности кристаллов. Это подтверждает и вид спектров сенсибилизированной фосфоресценции для всех исследуемых соединений, который имеет диффузный характер.
Итак, исследование влияния растворителя на характер температурной зависимости сенсибилизированной фосфоресценции показали, что растворитель приводит к изменениям величины наблюдаемого эффекта, что обусловлено различием локальной концентрации примесей в межблочном пространстве в зависимости от удобства растворителя. Т.е. влияние растворителя свелось к влиянию концентрации на характер температурной зависимости сенсибилизированной фосфоресценции.
Таким образом, результаты исследования влияния концентрации на параметры сенсибилизированной фосфоресценции указывают на то, что аномальный характер температурной зависимости I/I0 сенсибилизированной фосфоресценции наблюдается только для тех концентраций примесей, для которых характерно концентрационное тушение триплетных молекул. Поэтому можно предположить, что причиной увеличения концентрации триплетных молекул акцептора в температурной области 2 является снятие концентрационного тушения. Для выяснения механизмов снятия концентрационного тушения необходимо знать его природу. В зависимости от механизмов концентрационного тушения и процессов, приводящих к его снятию, увеличение интенсивности сенсибилизированной фосфоресценции в температурной области 2 может иметь обратимый или необратимый характер. Исследованию этого вопроса и посвящён следующий параграф.
3.3 НЕОБРАТИМЫЙ ХАРАКТЕР ХОДА КРИВОЙ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ КОНЦЕНТРАЦИИ ТРИПЛЕТНЫХ МОЛЕКУЛ АКЦЕПТОРА
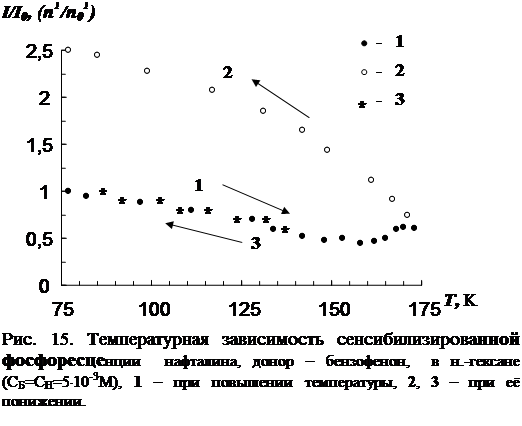 |
Одним из вопросов, которые необходимо было решить для выяснения природы процесса, приводящего к увеличению числа триплетных молекул в аномальной температурной области является обратимость этого процесса.
С этой целью был исследован характер изменения интенсивности сенсибилизированной фосфоресценции для донорно-акцепторных пар бензофенон-нафталин в н.-гексане (рис. 15) при повышении его температуры от 77 К до 175 К (кривая 1) и последующем охлаждении от 175 К до 77 К (кривая 2). Также было исследовано изменение интенсивности сенсибилизированной фосфоресценции в температурной области 1 для данной пары при изменении температуры раствора от 77 до 140 К и последующем его охлаждении до 77 К (кривая 3).
Как видно из рис. 15, увеличение интенсивности сенсибилизированной фосфоресценции в температурной области 2 имеет необратимый характер. При охлаждении образца от 175 К до 77 К тушение сенсибилизированной фосфоресценции, которое снимается в температурной области 2, не восстанавливается. Это указывает на необратимость процесса снятия концентрационного тушения. Если же образец нагреть до 150 К и затем его охладить до 77 К (кривая 3), то экспериментальные точки в зависимости интенсивности сенсибилизированной фосфоресценции от температуры укладываются на одну и ту же кривую. Это указывает на то, что изменение интенсивности сенсибилизированной фосфоресценции при нагревании до любой температуры из температурной области 1 является обратимым.
Для окончательного решения вопроса о необратимом характере процесса снятия концентрационного тушения в температурной области 2 был проведён следующий эксперимент. Исследовалась температурная зависимость интенсивности сенсибилизированной фосфоресценции аценафтена в н.-октане при нагревании раствора от 77 К до 185 К (рис.16, кривая 1), последующем его охлаждении от 185 К до 77 К (рис. 16, кривая 2) и повторном нагревании от 77 К до 180 К (рис. 16, кривая 3). Как и в случае нафталина в н.-гексане (рис. 15) изменение интенсивности сенсибилизированной фосфоресценции аценафтена при его нагревании от 77 К до 185 К имеет необратимый характер. Т.е. при последующем охлаждении зависимость имеет монотонный характер. Для одних и тех же температур интенсивность фосфоресценции при охлаждении раствора в несколько раз больше, чем при его нагревании. При последующем увеличении температуры от 77 К до 185 К процесс становится обратимым, на что указывает совпадение кривых 2 и 3. Последний результат говорит о полном снятии концентрационного тушения в температурной области 2, чем также подтверждает данную гипотезу.
Следует отметить, что для аценафтена в н.-октане увеличение интенсивности при 77 К после охлаждения раствора (кривая 2, рис. 16) превосходит первоначальное ее значение при этой же температуре (кривая 1, рис. 16) в 7.4 раза. Тогда как отношение этих величин для нафталина в н.-гексане при 77 К (кривые 1 и 2, рис.15) равно 2,5. Большее значение увеличения I/I0 в сравнении с нафталином в н.-гексане для аценафтена в н.-октане и следовало ожидать в данном случае, поскольку величина аномального температурного эффекта для первой системы больше.
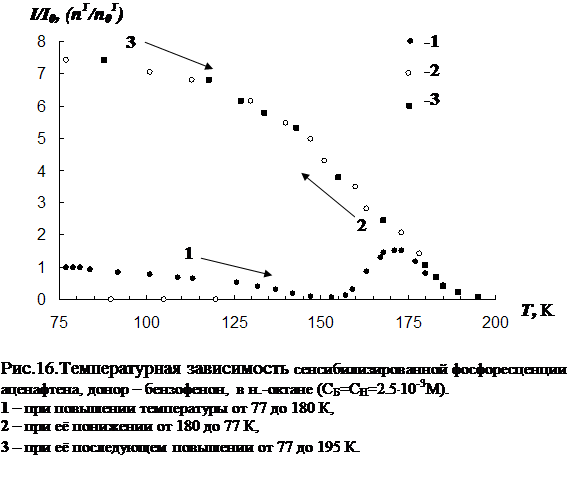 |
Теперь рассмотрим возможные причины, обуславливающие увеличение концентрации триплетных молекул акцептора.
Как было показано в 1.3 (формула 7), концентрация триплетных молекул может изменяться как за счёт относительной заселённости триплетного уровня b, так и в результате изменения общего числа молекул, участвующих в излучении N.
Запишем выражение (7) для случая сенсибилизированного заселения триплетного уровня:
![]() . (42)
. (42)
Здесь ![]() - концентрация триплетных
молекул акцептора,
- концентрация триплетных
молекул акцептора, ![]() - относительная
заселённость их триплетного уровня, NA
– число молекул акцептора, участвующих в излучении сенсибилизированной
фосфоресценции.
- относительная
заселённость их триплетного уровня, NA
– число молекул акцептора, участвующих в излучении сенсибилизированной
фосфоресценции.
С целью ответа на вопрос за счет изменения какого параметра происходит
изменение концентрации триплетных молекул акцептора, была определена (по
формуле (40)) относительная заселённость их триплетного уровня ![]() при 77 К для кривых 1 и 2
нафталина (рис. 15) и аценафтена (рис. 16). Эти результаты представлены в
таблице 6 и 7 соответственно.
при 77 К для кривых 1 и 2
нафталина (рис. 15) и аценафтена (рис. 16). Эти результаты представлены в
таблице 6 и 7 соответственно.
Таблица 6
ОТНОСИТЕЛЬНАЯ ЗАСЕЛЁННОСТЬ ТРИПЛЕТНОГО УРОВНЯ НАФТАЛИНА В Н.-ГЕКСАНЕ ПРИ 77 К.
tР, с
tT, с
![]() , отн.ед.
, отн.ед.
кривая 1
кривая 2
Таблица 7
ОТНОСИТЕЛЬНАЯ ЗАСЕЛЁННОСТЬ ТРИПЛЕТНОГО УРОВНЯ АЦЕНАФТЕНА В Н.-ОКТАНЕ ПРИ 77 К
tР, с
tТ, с
![]() , отн. ед.
, отн. ед.
кривая 1
кривая 2
Как видно из
табл. 6, значение ![]() уменьшается для
нафталина после снятия концентрационного тушения, тогда как концентрация его
триплетных молекул увеличивается в 2.5 раза. Для аценафтена (табл. 7) после
снятия концентрационного тушения величина
уменьшается для
нафталина после снятия концентрационного тушения, тогда как концентрация его
триплетных молекул увеличивается в 2.5 раза. Для аценафтена (табл. 7) после
снятия концентрационного тушения величина ![]() увеличивается
на 8 %, тогда как концентрация триплетных молекул увеличивается в 7.4 раза.
Таким образом, изменением
увеличивается
на 8 %, тогда как концентрация триплетных молекул увеличивается в 7.4 раза.
Таким образом, изменением ![]() нельзя
объяснить увеличение концентрации триплетных молекул акцептора. На основании
этого можно сделать вывод, что увеличение концентрации триплетных молекул, а
следовательно и интенсивность сенсибилизированной фосфоресценции после снятия
концентрационного тушения происходит за счёт увеличения числа молекул
акцептора, участвующих в излучении сенсибилизированной фосфоресценции.
нельзя
объяснить увеличение концентрации триплетных молекул акцептора. На основании
этого можно сделать вывод, что увеличение концентрации триплетных молекул, а
следовательно и интенсивность сенсибилизированной фосфоресценции после снятия
концентрационного тушения происходит за счёт увеличения числа молекул
акцептора, участвующих в излучении сенсибилизированной фосфоресценции.
3.4 ВЛИЯНИЕ СКОРОСТИ ЗАМОРАЖИВАНИЯ НА ПАРАМЕТРЫ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ
Поскольку характер изменения интенсивности сенсибилизированной фосфоресценции при повышении температуры зависит от предыстории образца, то представляло интерес сравнить параметры сенсибилизированной фосфоресценции при быстром и медленном замораживании.
Такие эксперименты были проведены для пары бензофенон-аценафтен в н.-гептане, концентрация донора и акцептора - 5×10-2 М. Результаты исследований положения максимума 0-0 полосы в спектре сенсибилизированной фосфоресценции lmax, времени разгорания tР и времени затухания tТ при различных способах замораживания приведены в таблице 8.
Таблица 8
Параметры сенсибилизированной фосфоресценции аценафтена, донор –бензофенон, в н.-гептане при различных условиях замораживания
| Условия замораживания |
lmax, нм |
tР, с |
tТ, с |
| Быстрое замораживание до 77 К | 481.4 | 1.35 | 2.10 |
| Быстрое замораживание + нагрев до 170 К и охлаждение до 77 К | 480.4 | 1.55 | 2.25 |
| Медленное замораживание до 77 К | 482.2 | 1.20 | 1.75 |
Из таблицы видно, что процесс увеличения числа молекул акцептора, участвующих в переносе энергии, происходящий при температурах из аномальной области 2 и медленное замораживание образца приводят к различным результатам. Первый процесс ведёт к смещению максимума в коротковолновую область, увеличению времени разгорания и времени затухания сенсибилизированной фосфоресценции. Такие же изменения параметров сенсибилизированной фосфоресценции характерны и для уменьшения концентрации примесей в растворе (табл.4). Медленное замораживание образца, как и увеличение концентрации молекул примесей в растворе приводит к смещению максимума 0-0 полосы в длинноволновую область и уменьшению времени разгорания и времени затухания сенсибилизированной фосфоресценции.
Такое поведение люминесцентных параметров при медленном замораживании образца следовало ожидать. Медленное замораживание, как и повышение концентрации, способствует образованию различных видов агрегатов (см. например [112]). К ним могут относиться, например, молекулы акцептора, внедрённые в кристаллы донора. Такие центры будут характеризоваться другими люминесцентными параметрами, отличными от мономерных молекул. Нами были исследованы люминесцентные характеристики молекул аценафтена, внедрённых в кристаллы бензофенона [28]. Положение максимума 0-0 полосы в спектре сенсибилизированной фосфоресценции – 482,5 нм, т.е. смещение наблюдается так же в длинноволновую область, как и в случае медленного замораживания.
Таким образом, можно сделать заключение, что медленное замораживание и процесс, происходящий в аномальной температурной области приводят к различным результатам. Так как в первом случае охлаждение происходит медленно, то, по-видимому, в процессе этого успевает произойти установление равновесия в системе (релаксация) при любой температуре. До достижения точки кристаллизации растворителя, при медленном замораживании, раствор более длительное время находится в жидком состоянии при низких температурах. Понижение температуры жидкости приводит к уменьшению растворимости, а это способствует образованию агрегатов. Другим отличием «предыстории» образца медленно замороженного от быстро замороженного является то, что он более длительное время пребывает в температурной области 2 в замороженном виде. Поэтому в нём также могут происходить процессы, приводящие к снятию концентрационного тушения. При нагревании такого раствора не наблюдается аномальных областей увеличения интенсивности сенсибилизированной фосфоресценции. Энергии теплового движения молекул при температурах ниже точки плавления растворителя не хватает для разрушения связей в агрегатах примесей. Поэтому в этом случае ход температурной кривой является обратимым.
При быстром замораживании не успевает произойти установления равновесия в системе, поэтому при 77 К фиксируется неравновесное состояние. Как показало медленное замораживание, процессы, ведущие в сторону равновесия в системе связаны с образованием агрегатов. Агрегаты представляют собой образования, состоящие из большого числа молекул примесей. Процесс их роста начинается с центров, состоящих из малого числа молекул примесей – ассоциатов. Если предположить, что при быстром замораживании происходит приостановка процесса агрегации на фазе ассоциатов, то дальнейшее поведение образца при повышении температуры можно прогнозировать следующим образом. Энергия связи для молекул в ассоциате является меньшей, чем в агрегатах или кристаллах. Энергии теплового движения молекул при температурах ниже точки плавления растворителя может оказаться достаточно, чтобы скорость процесса распада ассоциатов достигла наблюдаемых величин. Если скорость процесса распада станет сравнима со скоростью изменения температуры, то это должно сказаться на кривой зависимости интенсивности сенсибилизированной фосфоресценции от температуры.
3.5 ОСНОВНЫЕ РЕЗУЛЬТАТЫ ГЛАВЫ 3
Основные результаты данной главы можно представить следующим образом.
Увеличение интенсивности сенсибилизированной фосфоресценции в аномальной температурной области 2 происходит за счёт снятия концентрационного тушения сенсибилизированной фосфоресценции. В результате снятия концентрационного туше6ния происходит увеличение числа молекул акцептора энергии, участвующих в излучении сенсибилизированной фосфоресценции, тогда как относительная заселённость их триплетного уровня практически не изменяется. Этот процесс имеет необратимый характер, а следовательно не может быть обусловлен зависимостью переноса энергии от температуры [51].
На основании вышеизложенного и (40) можно предположить о наличии процессов в температурной области 2, приводящих к увеличению числа триплетных молекул акцептора, участвующих в излучении сенсибилизированной фосфоресценции с определённой энергией активации Еак. Тогда выдерживание образца при фиксированной температуре из аномальной области так же должно приводить к увеличению интенсивности сенсибилизированной фосфоресценции. Результаты таких исследований кинетики процесса при различных температурах приведены в следующей главе.
Глава 4
Влияние отжига на концентрацию триплетных молекул акцептора энергии
В предыдущей главе при обсуждении причин немонотонного характера температурной зависимости сенсибилизированной фосфоресценции примесных молекул в н.-парафиновых растворах было показано, что рост интенсивности связан не с зависимостью параметров переноса от температуры, а с повышением при данных температурах скорости какого-то физического процесса, приводящего к снятию концентрационного тушения триплетных состояний.
Поэтому необходимо было исследовать изменение люминесцентных характеристик донорно-акцепторных пар в результате выдерживания образца при постоянной температуре из аномальной области 2. Такое выдерживание образца при фиксированной температуре из аномальной области 2 в течение определённого времени в дальнейшем будем называть его отжигом.
В данной главе приведены результаты исследования влияния отжига на спектры, кинетику и интенсивность сенсибилизированной фосфоресценции молекул акцептора и обычной фосфоресценции молекул донора. На основании результатов этих исследований сделан вывод о том, что одной из причин концентрационного тушения возбужденных состояний примесных молекул в н.-парафиновых растворах является образование гетероассоциатов из молекул донора и акцептора, а причиной увеличения интенсивности фосфоресценции молекул донора и сенсибилизированной фосфоресценции акцептора в процессе отжига – их распад.
4.1 ИССЛЕДОВАНИЕ ВЛИЯНИЯ ОТЖИГА НА ИНТЕНСИВНОСТЬ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ И ХАРАКТЕР ЕЁ ТЕМПЕРАТУРНОЙ ЗАВИСИМОСТИ
Для исследования влияния отжига на параметры сенсибилизированной фосфоресценции были выбраны донорно-акцепторные пары бензофенон-аценафтен и бензофенон-нафталин в н.-октане и н.-декане. Выбор объектов в данном случае обусловлен существованием максимального эффекта увеличения концентрации триплетных молекул при нагревании раствора.
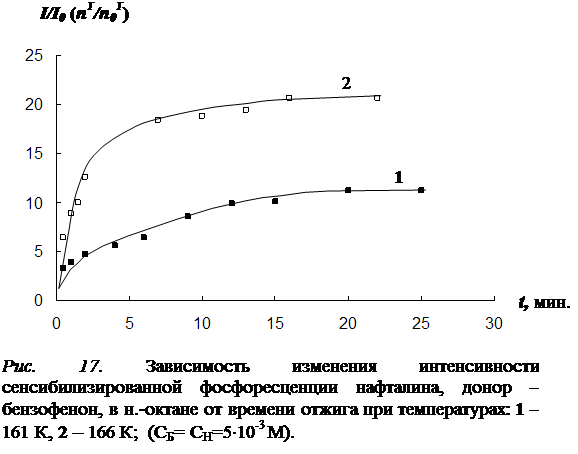 |
На рис. 17 приведена зависимость относительной интенсивности сенсибилизированной фосфоресценции I/I0 (nT/n0T) нафталина в н.-октане от времени отжига образца при двух фиксированных температурах: Т1 = 161 К (кривая 1) и Т2 = 166 К (кривая 2). Здесь I – интенсивность сенсибилизированной фосфоресценции охлажденного до 77 К после отжига в течение времени t образца, а I0 – интенсивность фосфоресценции неотожжённого образца при 77 К. Как видно из рисунка, интенсивность сенсибилизированной фосфоресценции, а следовательно и концентрация триплетных молекул нафталина растут с увеличением времени выдерживания образца при фиксированной температуре из области 2. Значение I/I0 (nT/n0T) с увеличением времени отжига монотонно стремится к предельному значению. Это указывает на то, что процесс, приводящий к увеличению числа триплетных молекул акцептора, участвующих в излучении сенсибилизированной фосфоресценции, характеризуется насыщением.
Максимальное значение, к которому стремится I/I0 (nT/n0T) зависит от температуры отжига. С увеличением температуры отжига максимальное значение I/I0 (nT/n0T) увеличивается. При температуре отжига Т1 = 161 К (кривая 1) после достижения насыщения значение I/I0 (nT/n0T) » 11, при Т2 = 166 К (кривая 2), максимальное значение I/I0 (nT/n0T) » 26. Кроме этого, как видно из рис. 12 от температуры отжига зависит также и скорость процесса, приводящего к росту интенсивности сенсибилизированной фосфоресценции нафталина. Скорость процесса тем больше, чем выше температура отжига.
Такие же исследования были проведены для пары бензофенон-аценафтен в н.-декане (рис. 18) при четырёх фиксированных температурах отжига: Т1 = 157 К (кривая 1), Т2 = 167 К (кривая 2), Т3 = 172 К (кривая 3) и Т4 = 177 К (кривая 4). Для данной пары, как и для предыдущей, интенсивность сенсибилизированной фосфоресценции, а следовательно и число триплетных молекул акцептора энергии I/I0 (nT/n0T) растет с увеличением времени отжига. Предельные значения, к которым стремится I/I0 (nT/n0T), и для данной пары зависят от температуры отжига. С повышением температуры отжига предельные значения I/I0 (nT/n0T) увеличиваются. Так, при температуре Т1 = 157 К отжиг приводит к максимальному значению I/I0 (nT/n0T) » 15, при Т2 = 167 К - I/I0 (nT/n0T) » 18, при Т3 = 172 К – I/I0 (nT/n0T) » 30 и при Т4 = 177 К – I/I0 (nT/n0T) » 35. Скорость процесса, приводящего к увеличению интенсивности сенсибилизированной фосфоресценции аценафтена так же возрастает с увеличением температуры отжига.
Существование насыщения I/I0 (nT/n0T) при отжиге говорит о том, что в растворе с прошествием времени достигается равновесное состояние. Это состояние в условиях стационарного возбуждения характеризуется определённым числом триплетных молекул акцептора энергии, участвующих в излучении. Причём число триплетных молекул акцептора, характеризующих равновесное состояние, растёт с увеличением температуры отжига.
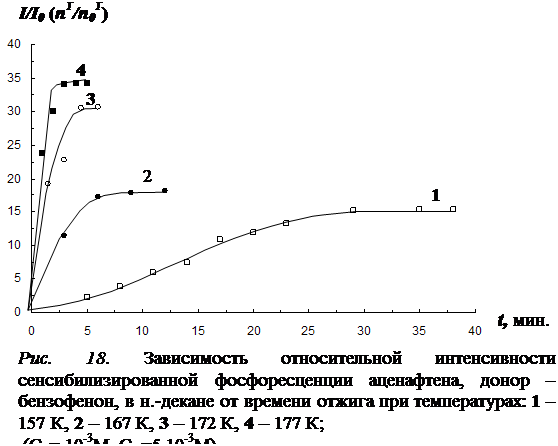 |
Таким образом, на основании этих результатов можно сделать вывод о том, что увеличение интенсивности сенсибилизированной фосфоресценции в температурной области 2 и его необратимый характер обусловлен отжигом образца в процессе его нагревания. Подтверждением этого вывода является то, что интенсивность сенсибилизированной фосфоресценции отожжённого образца монотонно убывает при повышении температуры (рис. 19, кривая 1).
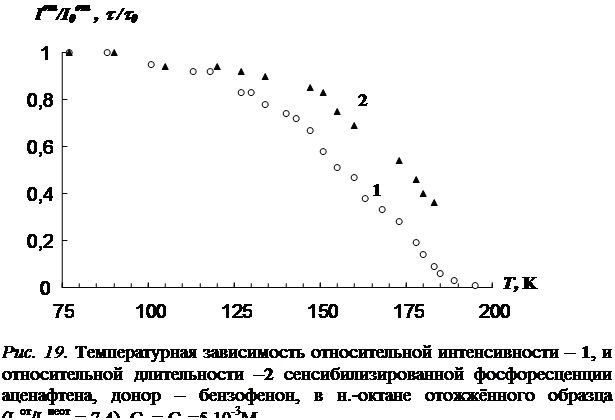 |
Уменьшение интенсивности сенсибилизированной фосфоресценции отожжённого образца при повышении температуры сопровождается падением времени затухания tТ (кривая 2, рис. 19). Это и монотонный характер температурной зависимости интенсивности сенсибилизированной фосфоресценции отожжённого образца указывают на то, что в системе остаётся только один процесс, влияющий на концентрацию триплетных молекул акцептора энергии – динамическое тушение.
Эти результаты дают основание утверждать, что отжиг снимает концен
трационное тушение сенсибилизированной фосфоресценции органических молекул в замороженных н.-парафиновых растворах.
Представляло интерес исследовать влияние отжига на концентрационную зависимость интенсивности сенсибилизированной фосфоресценции. С этой целью был проведён следующий эксперимент.
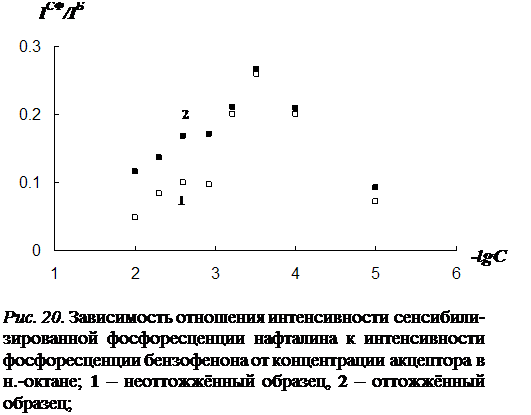 |
Для растворов, в которых концентрация молекул донора (бензофенона) оставалась постоянной и равнялась 10-3 М, а концентрация акцептора (нафталина) изменялась от 10-5 М до 10-2 М, измерялось отношение интенсивности сенсибилизированной фосфоресценции акцептора к интенсивности фосфоресценции донора IСФ/IБ (рис.20). Измерения IСФ/IБ производились до отжига образца (кривая 1) и после его отжига (кривая 2) при 185 К по достижению насыщения.
Величина IСФ/IБ для неотожжённого образца при увеличении концентрации нафталина от 10-5 до 5×10-4 М линейно возрастает. Эти данные говорят о том, что в настоящем диапазоне не наблюдается концентрационного тушения сенсибилизированной фосфоресценции. Экспериментальные точки кривой 2 отожжённого образца здесь практически совпадают с точками кривой 1. Следовательно, в области прямой положительной зависимости IСФ/IБ от логарифма концентрации акцептора, низкотемпературный отжиг практически не влияет на число триплетных молекул акцептора.
Далее, при повышении концентрации от 5×10-4 до 10-2 М для неотожжённого образца наблюдается резкое уменьшение IСФ/IБ – область концентрационного тушения сенсибилизированной фосфоресценции. Здесь отжиг приводит к увеличению значений IСФ/IБ по сравнению с кривой 1. Т.е. экспериментальные данные свидетельствуют о снятии, хотя и не полном, концентрационного тушения сенсибилизированной фосфоресценции в процессе отжига.
Итак, как видно из результатов настоящего опыта, отжиг приводит к увеличению интенсивности сенсибилизированной фосфоресценции только в тех случаях, когда наблюдается ее концентрационное тушение. При этом отжиг приводит только к частичному снятию концентрационного тушения сенсибилизированной фосфоресценции.
Следует отметить, что обезгаживание образца путём многократной перекристаллизации в вакууме не изменяло влияние отжига на интенсивность сенсибилизированной фосфоресценции. Это даёт основание полагать, что снятие тушения сенсибилизированной фосфоресценции в процессе отжига не связано с наличием кислорода в растворе.
4.2 ВЛИЯНИЕ ОТЖИГА НА СПЕКТРЫ И КИНЕТИКУ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ
Если в результате отжига замороженного н.-парафинового раствора органических соединений снимается концентрационное тушение сенсибилизированной фосфоресценции, то можно ожидать, что при этом параметры спектров и кинетики сенсибилизированной фосфоресценции будут изменяться также, как при понижении концентрации раствора. С целью проверки этого предположения было исследовано влияние отжига на спектры сенсибилизированной фосфоресценции нафталина (рис. 21) и аценафтена (рис. 22) в н.-октане и кинетику сенсибилизированной фосфоресценции аценафтена (табл. 10 и 11).
 |
На рис. 21 приведены спектры фосфоресценции пары бензофенон-нафталин в н.-октане для неотожжённого (а) и отожжённого (б) образцов. Возбуждался только донор энергии. Цифрами 1 на рисунке обозначены линии, принадлежащие спектру фосфоресценции бензофенона, цифрами 2 – спектру сенсибилизированной фосфоресценции нафталина.
Как видно из рис. 21, в результате отжига наблюдается увеличение интенсивности сенсибилизированной фосфоресценции. При этом также наблюдается и увеличение интенсивности фосфоресценции молекул донора. Однако, увеличение интенсивности фосфоресценции молекул донора при этом происходит в меньшее число раз. Так, интенсивность фосфоресценции бензофенона после отжига увеличивается в 4 раза, а интенсивность сенсибилизированной фосфоресценции нафталина увеличивается после отжига в 40 раз.
В результате отжига раствора происходит также смещение максимума 0-0 полосы lmax в коротковолновую область на 40-50 см-1. Для неотожжённого образца lmax – 473.0 нм, для отожжённого – 472.0 нм.

На рис.
22 приведён спектр фосфоресценции пары бензофенон-аценафтен в н.-октане для
неотожжённого образца (а) и для отожжённого в течение 4 мин. при 180 К (б). Как
видно, и в этом случае отжиг приводит к увеличению как интенсивности
фосфоресценции донора, так и интенсивности сенсибилизированной фосфоресценции
акцептора. Рост интенсивности фосфоресценции донора при этом меньше, чем
акцептора. При этом так же наблюдается смещение максимума 0-0 полосы в спектре
фосфоресценции аценафтена в коротковолновую область на 40-50 см –1.
Таким образом, сравнение спектральных характеристик неотожжённого и отожжённого образцов показало, что отжиг приводит к смещению максимума 0-0 полосы в спектре сенсибилизированной фосфоресценции в коротковолновую область, а так же к увеличению интенсивности свечения как акцептора, так и донора энергии.
Изучение влияния отжига на кинетические характеристики сенсибилизированной фосфоресценции, в частности на время затухания tТ, производилось на паре бензофенон-аценафтен в н.-октане. Результаты этих измерений приведены в табл. 12.
Таблица 12.
Параметры сенсибилизированной фосфоресценции аценафтена различной концентрации (СА) до и после отжига.
(Отжиг производился в течение 5 мин. при 175 К; в качестве донора использовался бензофенон неизменной концентрации – СБ = 5×10-3 М; растворитель – н.-октан.)
|
СА, М |
Iот/Iнеот |
lmнеот, нм |
lmот, нм |
tТнеот, с |
tТот, с |
|
5×10-3 |
7.5 | 481.5 | 480.5 | 1.65 | 2.50 |
|
10-3 |
1.3 | 481.0 | 480.0 | 2.40 | 2.65 |
|
5×10-4 |
1.1 | 480.5 | 480.0 | 2.60 | 2.70 |
Для исследования использовались различные концентрации акцептора энергии: 5×10-3 М – из области, где наблюдается сильное концентрационное тушение сенсибилизированной фосфоресценции, 10-3 и 5×10-4 М – из области, где тушение уменьшается. Концентрация донора не изменялась и была равна 5×10-3 М. Отжиг производился во всех случаях при температуре 175 К, в течение 5 минут. Такие время и температура отжига, с точки зрения предварительных оценочных экспериментов, приводят к максимальному значению интенсивности по достижению насыщения за указанный промежуток времени.
В столбце 2 табл. 12 приведено отношение интенсивности сенсибилизированной фосфоресценции после отжига Iот к его значению до отжига Iнеот при заданной концентрации акцептора. Данные опыта свидетельствуют о максимальном росте интенсивности сенсибилизированной фосфоресценции после отжига при концентрации 5×10-3 М. Концентрации 10-3 и 5×10-4 М после отжига дают незначительное увеличение интенсивности.
В 3 и 4 столбцах
таблицы указаны положения максимумов 0-0 полосы в спектре сенсибилизированной
фосфоресценции аценафтена до (lmнеот) и после (lmот) отжига. При
всех рассмотренных концентрациях после отжига максимум смещается в сторону коротких
длин волн. В первом случае это смещение составляет 10 ![]() , в двух последующих – 5
, в двух последующих – 5 ![]() . Если проследить изменение
положения максимума по столбцам, при понижении концентрации акцептора, то
очевидно аналогичное поведение системы. Уменьшение концентрации так же ведёт к
коротковолновому смещению максимума.
. Если проследить изменение
положения максимума по столбцам, при понижении концентрации акцептора, то
очевидно аналогичное поведение системы. Уменьшение концентрации так же ведёт к
коротковолновому смещению максимума.
В двух последних столбцах представлены значения времени затухания сенсибилизированной фосфоресценции неотожжённого - tТнеот и отожжённого - tТот образцов. Для каждого из рассмотренных случаев после отжига наблюдается более медленное затухание свечения. При концентрации 5×10-3 М увеличение времени затухания после отжига происходит на величину 0.85 с и составляет наибольшее значение в данном опыте, при 10-3 М tТот увеличивается на 0.15 с по сравнению с tТнеот, при 5×10-4 М – на 0.1 с. Если проследить поведение системы относительно данного параметра при понижении концентрации акцептора, то можно увидеть подобные результаты: уменьшение концентрации акцептора ведёт к увеличению времени затухания сенсибилизированной фосфоресценции.
Таким образом, исследования спектральных и кинетических характеристик сенсибилизированной фосфоресценции неотожжённого и отожжённого образцов при различных концентрациях показали, что процесс низкотемпературного отжига и уменьшение концентрации акцептора в растворе приводят к одинаковым результатам, а именно:
1) увеличению интенсивности (для области, где наблюдается концентрационное тушение);
2) коротковолновому смещению максимума 0-0 полосы;
3) увеличению времени затухания.
С целью установления причин увеличения интенсивности сенсибилизированной
фосфоресценции примесных молекул в данных системах в результате отжига образца
была определена относительная заселённость триплетного уровня молекул акцептора
![]() до и после отжига.
Результаты этих исследований представлены в табл. 13. Здесь же приведены
результаты определения константы перехода kП
молекул аценафтена из основного в триплетное состояние в различных
растворителях.
до и после отжига.
Результаты этих исследований представлены в табл. 13. Здесь же приведены
результаты определения константы перехода kП
молекул аценафтена из основного в триплетное состояние в различных
растворителях.
Таблица 13.
Параметры, характеризующие молекулы аценафтена в условиях переноса возбуждения, донор – бензофенон, до и после отжига.
|
Растворитель |
СБ, М |
СА, М |
|
kП, с-1 |
Iот/Iнеот |
||
| неотож. | отож. | неотож. | отож. | ||||
| н.-октан |
5×10-3 |
5×10-3 |
0.46 | 0.50 | 0.49 | 0.42 | 10 |
| н.-гептан |
5×10-2 |
5×10-2 |
0.38 | 0.32 | 0.29 | 0.21 | 2 |
| н.-гексан |
10-2 |
10-2 |
0.35 | 0.33 | 0.25 | 0.21 | 2 |
Относительная погрешность при определении ![]() и
kП составляла не более 10 %.
и
kП составляла не более 10 %.
Как видно из таблицы, разница в значениях ![]() и
kП для отожжённого и неотожжённого
образца не превышает ошибки измерения, тогда как интенсивность сенсибилизированной
фосфоресценции в результате отжига увеличивается в несколько раз. С учётом
формулы (40) можно сделать вывод, что увеличение интенсивности
сенсибилизированной фосфоресценции в результате отжига происходит не за счёт
изменения
и
kП для отожжённого и неотожжённого
образца не превышает ошибки измерения, тогда как интенсивность сенсибилизированной
фосфоресценции в результате отжига увеличивается в несколько раз. С учётом
формулы (40) можно сделать вывод, что увеличение интенсивности
сенсибилизированной фосфоресценции в результате отжига происходит не за счёт
изменения ![]() , а за счёт увеличения
числа молекул акцептора NA,
участвующих в излучении сенсибилизированной фосфоресценции.
, а за счёт увеличения
числа молекул акцептора NA,
участвующих в излучении сенсибилизированной фосфоресценции.
Для того, чтобы выяснить, за счёт каких процессов происходит увеличение числа молекул акцептора, участвующих в излучении сенсибилизированной фосфоресценции, необходимо было также изучить влияние отжига на параметры фосфоресценции молекул донора в присутствие молекул акцептора в растворе. Результаты этих исследований приведены в следующем параграфе.
4.3 ВЛИЯНИЕ ОТЖИГА НА ПАРАМЕТРЫ ФОСФОРЕСЦЕНЦИИ ДОНОРА ЭНЕРГИИ
Как отмечалось в предыдущем параграфе, интенсивность фосфоресценции донора в присутствие молекул акцептора в растворе после отжига также возрастает.
Для случая (рис. 21), когда акцептором энергии является нафталин, интенсивность фосфоресценции бензофенона возрастает в н.-октане для отожжённого образца в 4 раза в сравнении с неотожженным. Когда акцептором энергии является аценафтен, (рис. 22), интенсивность фосфоресценции бензофенона возрастает в 2 раза. В обоих случаях концентрация бензофенона была равна концентрации молекул акцептора и равна 5×10-3 М.
Увеличение интенсивности фосфоресценции молекул донора в присутствие молекул акцептора в результате отжига образца указывает на то, что отжиг снимает также тушение триплетных состояний молекул донора.
Следует заметить, что интенсивность фосфоресценции молекул донора в результате отжига образца увеличивается всегда в меньшее число раз, чем интенсивность сенсибилизированной фосфоресценции молекул акцептора. Одной из возможных причин этого может быть то, что на интенсивности фосфоресценции донора сказывается только тушение их триплетных состояний, а на интенсивность сенсибилизированной фосфоресценции – как тушение триплетных молекул донора, так и тушение триплетных молекул акцептора.
С целью выяснения причин возрастания интенсивности фосфоресценции молекул донора была исследована кинетика затухания его фосфоресценции в присутствие молекул акцептора для неотожжённого и отожжённого образцов, а так же кинетика затухания его фосфоресценции в отсутствие молекул акцептора. Результаты представлены в табл. 14.
Таблица 14.
Время затухания фосфоресценции бензофенона при 77 К
|
|
в отсутствие аценафтена | в присутствии аценафтена до отжига | в присутствии аценафтена после отжига |
|
|
6.1 | 3.9 | 5.0 |
|
|
164 | 256 | 200 |
|
kТ, с-1 |
0 | 92 | 36 |
|
I/I0 |
1 | 0.2 | 0.4 |
В качестве акцептора энергии использовался аценафтен, в качестве растворителя – н.-октан. Концентрация аценафтена в растворе равнялась концентрации бензофенона и составляла 5×10-3 М. Отжиг образца производился в течение 4 мин. при Т = 180 К.
Как видно из таблицы, при добавлении аценафтена в раствор бензофенона в н.-октане, время затухания фосфоресценции последнего уменьшается от 6.1 мс до 3.9 мс. Отжиг этого образца приводит к увеличению интенсивности фосфоресценции бензофенона в 2 раза (рис. 22), при этом время затухания увеличивается в 1.3 раза. Одновременное увеличение интенсивности и времени затухания фосфоресценции бензофенона может быть обусловлено только снятием тушения его триплетных состояний. Однако, это не может быть связано с уменьшением тушения триплетных молекул донора в результате переноса энергии на одиночные молекулы акцептора, участвующие в излучении сенсибилизированной фосфоресценции, поскольку её интенсивность при этом также возрастает. На основании этого можно утверждать о наличии других каналов тушения триплетных молекул донора в присутствие молекул акцептора, кроме переноса энергии на одиночные молекулы акцептора, участвующие в излучении сенсибилизированной фосфоресценции. Эти каналы дезактивации энергии триплетного возбуждения молекул донора назовём дополнительными каналами тушения. С их учётом время затухания фосфоресценции донора можно представить в следующем виде:
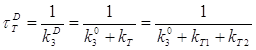 . (43)
. (43)
Здесь ![]() - константа перехода
молекул донора из триплетного состояния в основное в отсутствии молекул
акцептора в растворе;
- константа перехода
молекул донора из триплетного состояния в основное в отсутствии молекул
акцептора в растворе;
kT1 - константа перехода молекул донора из триплетного состояния в основное в результате передачи энергии молекулам акцептора, участвующих в излучении сенсибилизированной фосфоресценции;
kT2 - константа перехода молекул донора из триплетного состояния в основное в результате дополнительного тушения.
Отжиг раствора снимает дополнительное тушение. Однако, как отмечалось
выше, увеличение интенсивности опережает изменение ![]() . Если бы изменение
интенсивности было обусловлено только одним процессом, в результате которого
также изменяется
. Если бы изменение
интенсивности было обусловлено только одним процессом, в результате которого
также изменяется ![]() , то
отношение интенсивностей до и после отжига не превышало бы отношение времен затухания
[19]
, то
отношение интенсивностей до и после отжига не превышало бы отношение времен затухания
[19]
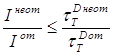 . (44)
. (44)
Поэтому можно
предположить, что существует два типа дополнительного тушения. В результате
одного из них происходит уменьшение ![]() ,
а в результате второго происходит полное тушение триплетных состояний
определённой части молекул донора. Молекулы донора, испытывающие тушение
второго типа, не участвуют в излучении, и поэтому это тушение не влияет на
,
а в результате второго происходит полное тушение триплетных состояний
определённой части молекул донора. Молекулы донора, испытывающие тушение
второго типа, не участвуют в излучении, и поэтому это тушение не влияет на ![]() .
.
В результате
отжига снимается оба типа тушения. Снятие дополнительного тушения типа I приводит к
увеличению ![]() , а
следовательно и интенсивности за счёт изменения относительной заселённости
триплетного уровня, а снятие дополнительного тушения типа II – только к
увеличению интенсивности за счёт увеличения общего числа молекул донора,
участвующих в излучении.
, а
следовательно и интенсивности за счёт изменения относительной заселённости
триплетного уровня, а снятие дополнительного тушения типа II – только к
увеличению интенсивности за счёт увеличения общего числа молекул донора,
участвующих в излучении.
Увеличение интенсивности фосфоресценции донора в результате отжига позволяет отбросить из рассмотрения следующий возможный механизм увеличения числа молекул акцептора энергии, участвующих в излучении сенсибилизированной фосфоресценции. В процессе отжига образца система из термодинамически неустойчивого состояния переходит в более устойчивое, которое соответствует более равномерному распределению молекул примеси. В результате чего часть молекул акцептора, которые ранее не участвовали в переносе энергии, попадают в сферу обменных взаимодействий с молекулами донора и теперь участвуют в излучении. Очевидно, что при этом должно усиливаться тушение триплетных молекул донора энергии, в результате чего интенсивность и время затухания их фосфоресценции должны уменьшиться. Что противоречит экспериментальным результатам, приведенным выше.
4.4 ИССЛЕДОВАНИЕ ЗАКОНА НАКОПЛЕНИЯ ЧИСЛА МОЛЕКУЛ АКЦЕПТОРА, УЧАСТВУЮЩИХ В ИЗЛУЧЕНИИ, В ПРОЦЕССЕ ОТЖИГА
В 4.2 было показано, что увеличение интенсивности сенсибилизированной фосфоресценции в результате отжига образца происходит за счёт увеличения числа молекул акцептора, участвующих в излучении. Поэтому для выяснения физической природы процесса, приводящего к увеличению общего числа молекул акцептора, участвующих в излучении, необходимо было изучить закон их накопления в процессе отжига.
Обозначим интенсивность сенсибилизированной фосфоресценции после быстрого замораживания образца до 77 К через I(0). После отжига образца в течение определённого времени t при температуре Т и последующем охлаждении до 77 К интенсивность сенсибилизированной фосфоресценции обозначим через I(t). Тогда DI(t) = I(t) – I(0) – означает прирост интенсивности сенсибилизированной фосфоресценции в процессе отжига образца в течение этого времени.
По характеру кривых зависимостей относительной интенсивности сенсибилизированной фосфоресценции от времени отжига (рис. 17, 18) можно предположить, что при фиксированной температуре Т прирост интенсивности DI(t) в зависимости от времени отжига происходит по закону, определяемому экспонентой:
DI(t) = DI(¥){1-exp(-t/t)}, (45)
с характерным временем t, которое зависит от температуры отжига. DI(¥) - прирост интенсивности при длительном отжиге образца - t » t.
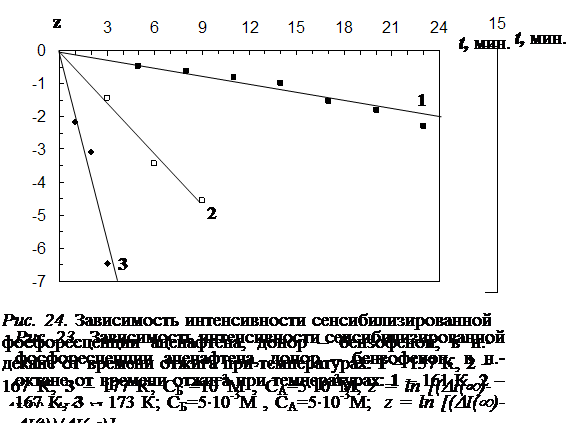 |
Экспериментально эта зависимость была проверена для пар – бензофенон-аценафтен в н.-октане и н.-декане и бензофенон-нафталин в н.-гексане, н.-октане и н.-декане. На рис. 23-27 в указанном порядке для данных соединений представлены графики зависимости [DI(t) - DI(¥)]/DI(¥) от t в полулогарифмическом масштабе.
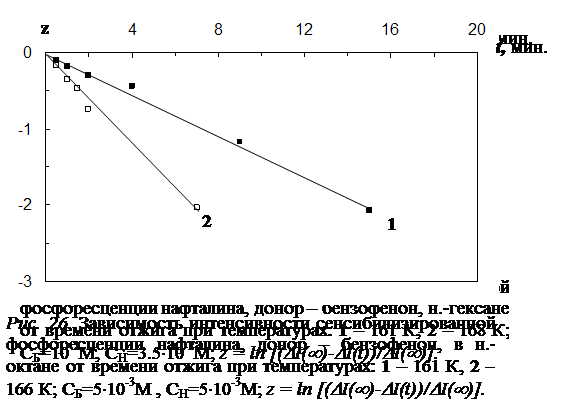 |
Как видно из рисунков, экспериментальные точки хорошо укладываются на экспоненту (сплошная линия) с различными углами наклона, определяемыми температурой отжига. Ве личина, обратная тангенсу угла наклона прямых, соответствует характерному времени t процесса при данной температуре отжига.
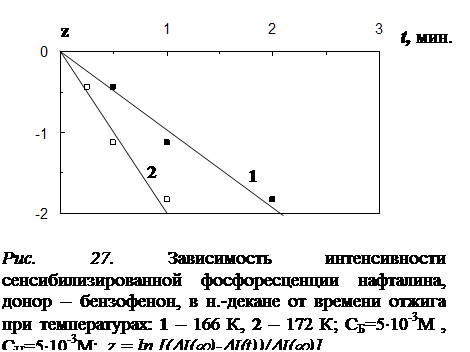 |
В табл. 15 приведены значения t , определённые из представленных на рис. 23-27 зависимостей.
Как видно из таблицы, для всех исследованных систем повышение температуры отжига раствора приводит к уменьшению характерного времени процесса нарастания.
Таким образом, на основании этих экспериментальных данных можно утверждать, что прирост стационарной интенсивности сенсибилизированной фосфоресценции в процессе отжига хорошо описывается экспонентой (39) с характерным временем t, которое уменьшается при повышении температуры отжига.
Поскольку, как отмечалось выше, в отсутствие реабсорбции излучения интенсивность сенсибилизированной фосфоресценции I(t) пропорциональна концентрации триплетных молекул акцептора энергии nT(t), то для последних также можно записать:
DnT(t) = DnT(¥){1 - exp(-t/t)}, (46)
где DnT(t)- изменение концентрации триплетных молекул нафталина за время отжига t.
Таблица 15.
Характерное время t процесса нарастания числа одиночных молекул акцептора, участвующих в переносе энергии в процессе отжига.
|
Соединение |
РастворительТемпература отжига, К |
t, мин. |
||
|
Бензофенон + аценафтен |
н.-октан |
СБ = 5×10-3 М СА = 5×10-3 М |
161 | 3.06 |
| 167 | 0.99 | |||
| 173 | 0.38 | |||
| н.-декан |
СБ = 10-3 М СА = 5×10-3 М |
157 | 9.81 | |
| 167 | 1.93 | |||
| 177 | 0.47 | |||
|
Бензофенон + нафталин |
н.-гексан |
СБ = 10-2 М СН = 3.5×10-3 М |
161 | 4.65 |
| 168 | 1.87 | |||
| н.-октан |
СБ = 5×10-3 М СН = 5×10-3 М |
161 | 7.41 | |
| 166 | 3.54 | |||
| н.-декан |
СБ = 5×10-3 М СН = 5×10-3 М |
166 | 1.12 | |
| 172 | 0.56 |
В 4.2 было
показано, что изменение концентрации триплетных молекул акцептора в процессе
отжига сопровождается практически неизменной относительной заселённостью
триплетного уровня - ![]() .
Основываясь на выражении (42) было сделано заключение, что изменение DnT(t) происходит за
счёт снятия концентрационного тушения. Поэтому аналогичный (46) закон
характеризует и рост числа мономерных молекул акцептора, участвующих в переносе
энергии.
.
Основываясь на выражении (42) было сделано заключение, что изменение DnT(t) происходит за
счёт снятия концентрационного тушения. Поэтому аналогичный (46) закон
характеризует и рост числа мономерных молекул акцептора, участвующих в переносе
энергии.
Таким образом, для прироста в процессе отжига общего числа молекул акцептора, участвующих в переносе энергии можно записать:
Dn(t) = Dn(¥){1-exp(-t/t)}. (47)
Величина, обратная t, характеризует скорость прироста при данной температуре концентрации триплетных молекул акцептора энергии, q = 1/t , и называется константой скорости процесса [161].
Итак, прирост в результате отжига образца числа молекул, участвующих в излучении сенсибилизированной фосфоресценции происходит по экспоненциальному закону. Константа скорости этого процесса зависит от температуры. В дальнейшем необходимо было определить характер зависимости константы скорости указанного выше процесса от температуры.
4.5 ОПРЕДЕЛЕНИЕ ЭНЕРГИИ АКТИВАЦИИ ПРОЦЕССА, ПРИВОДЯЩЕГО К УВЕЛИЧЕНИЮ ЧИСЛА МОЛЕКУЛ АКЦЕПТОРА, УЧАСТВУЮЩИХ В ИЗЛУЧЕНИИ
Возможными причинами увеличения после отжига концентрации молекул акцептора, участвующих в излучении, являются либо диффузионные процессы, приводящие к перераспределению имеющихся в растворе до отжига мономерных молекул, либо повышение числа мономерных молекул в растворе в результате распада более сложных образований. Хотя не исключены и другие процессы.
Выше было показано, что в твёрдом теле подобные физические и химические процессы обычно характеризуются Аррениусовской зависимостью константы скорости процесса от температуры:
q(Т) = q(¥) ехр (-Еак/RT) (48)
где q(¥) - предэкспоненциальный множитель, Еак- энергия активации процесса.
Соответственно для t :
t(Т) = (1/ q(¥)) ехр (Еак/RT). (49)
Представляло интерес экспериментально проверить эту зависимость.
Прологарифмируем уравнение Аррениуса (48):
lnt = Еак/RT- ln [q(¥)]. (50)
Написав это уравнение для различных температур Т1 и Т2 и вычтя второе уравнение из первого, получим:
ln(t1/t2)= Еак/R (1/T1- 1/Т2). (51)
Если это уравнение справедливо, то на графике в координатах ln(t1/t) от (1/T1- 1/Т) экспериментальные точки должны располагаться на прямой с тангенсом угла наклона, равным Еак/R.
На рис. 28 представлена данная зависимость для пар бензофенон-аценафтен в н.-октане (а), в н.-декане (б) и бензофенон-нафталин в н.-декане (в). Как видно из рисунка, экспериментальные точки хорошо укладываются на экспоненту (сплошная линия). Это говорит об экспоненциальной зависимости характеристического времени процесса t от температуры. Следовательно, и константа скорости q физического процесса, происходящего при отжиге экспоненциально растёт с повышением температуры.
Таким образом, на основании этих экспериментальных данных можно утверждать, что физический процесс, приводящий к увеличению числа участвующих в переносе энергии мономерных молекул акцептора при отжиге описывается Аррениусовской зависимостью константы скорости процесса от температуры.
Величина тангенса угла наклона прямых q позволяет определить энергию активации процесса: Еак= R tgq.
В табл. 16 представлены результаты определения энергии активации процесса для пар бензофенон-аценафтен и бензофенон-нафталин в различных растворителях. По порядку величины энергия активации процесса для исследованных ароматических углеводородов находится в пределах от 29 до 45 кДж/моль (от 0.30 до 0.47 эВ). Ошибка измерения этой величины составляла » 30 %.
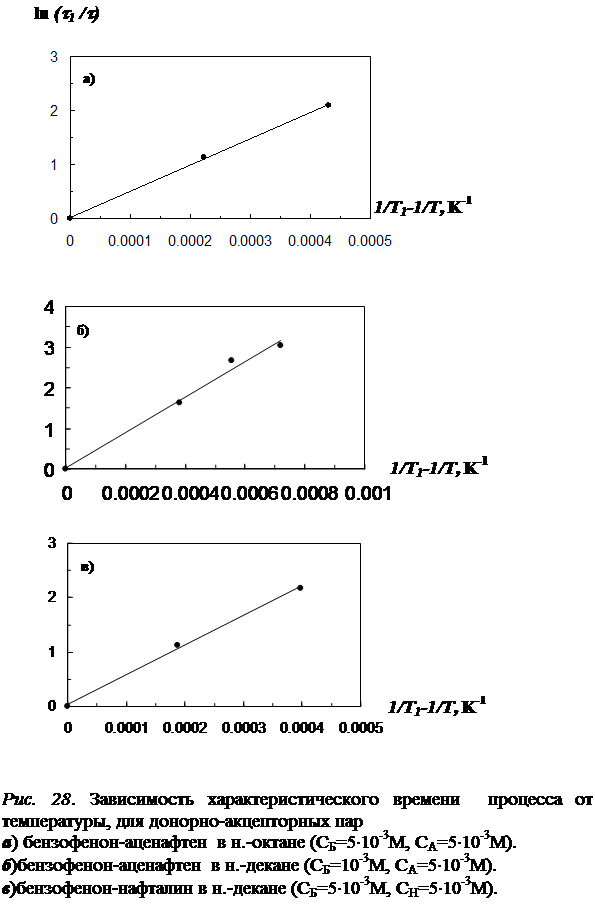
Таблица 16.
Энергия активации процесса увеличения общего числа молекул акцептора, участвующих в переносе энергии