Реферат: Дискриминация гипотез по кинетическим экспериментам
Раздел: Рефераты по химии
Тип: реферат
Дискриминация гипотез по кинетическим экспериментам
Кинетические методы используют для проверки адекватности гипотез, оставшихся после других методов дискриминации, решая обратную задачу химической кинетики – оценивание констант и сравнение полученных кинетических моделей статистическими методами. Вместе с тем, весьма эффективными путями использования кинетического эксперимента, как было отмечено выше, являются измерения кинетических изотопных эффектов (КИЭ) и анализ селективности стехиометрически многозначных процессов в зависимости от концентраций реагентов и от величины степени превращения XA.
Кинетические изотопные эффекты
Первичным кинетическим изотопным эффектом называют различие в скоростях разрыва (образования) химических связей при замене одного из атомов рвущейся (образующейся) связи его изотопом.
В основе первичного КИЭ при разрыве или образовании связи, например, С-Н, лежит различие величин нулевой энергии колебательного уровня этой связи, зависящей от природы изотопов, например, С (12С, 13С, 14С) или Н (1Н, 2D, 3T). Так, нулевая энергия связи U0 C-H ~ на 5 кДж/моль выше U0 для С-D. Поскольку соответствующие частоты колебаний в переходном состоянии в значительно меньшей степени зависят от массы изотопа (при наличии других тяжелых атомов и групп в реагентах), энергия активации в случае более легкого изотопа (С-Н) будет также ниже энергии активации для С-D и С-Т. Максимальные КИЭ разрыва связи без учета образования новых связей составляют:
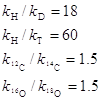
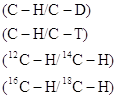
Поскольку в переходном состоянии связь рвется не полностью, максимальные значения КИЭ не достигаются. Величина КИЭ будет тем больше, чем в большей степени связи C-X, M-X, M-H разрываются в переходном состоянии.
Из теоретического анализа простой реакции переноса протона
![]()
в апротонных растворителях сделаны следующие выводы:
1) Основной вклад в КИЭ по водороду вносит потеря энергии валентного колебания связи А–Н при образовании переходного состояния. Частоты валентных колебаний в переходном состоянии ниже, чем в начальном и в меньшей степени зависят от массы изотопа.
2) Величина КИЭ определяется главным образом разностью энергий активации, а не отношением предэкспонентов.
3) Значения КИЭ обычно меньше, чем это следует из значений нулевых частот колебаний связей А–Н и A–D.
4) При использовании трех изотопов 1H, 2D и 3Т выполняется соотношение
![]()
Если связь в реагенте рвется в лимитирующей стадии, можно ожидать при замене, например H на D, значительного КИЭ. Так, в реакции
![]()
КИЭ ![]() . Это означает, что именно связь
C-D рвется в лимитирующей стадии. В квазиравновесных стадиях наблюдаются
равновесные (термодинамические) изотопные эффекты (ТИЭ).
. Это означает, что именно связь
C-D рвется в лимитирующей стадии. В квазиравновесных стадиях наблюдаются
равновесные (термодинамические) изотопные эффекты (ТИЭ).
Пример. При исследовании реакции окисления этилена хлоридом Pd(II) (Вакер-процесс) было обнаружено, что в реакциях
![]()
![]()
![]() и все четыре атома D переходят в
ацетальдегид. Следовательно, С-Н связь не рвется в лимитирующей стадии. Вместе
с тем, при замене H на D в воде
и все четыре атома D переходят в
ацетальдегид. Следовательно, С-Н связь не рвется в лимитирующей стадии. Вместе
с тем, при замене H на D в воде ![]() . Этот эффект, судя по кинетике
процесса есть ТИЭ, появление которого связано с разницей констант автопротолиза
Kw D2O и H2O (Kw H2O в 5.13 раз выше Kw D2O), и, соответственно, констант
кислотной диссоциации аквакомплексов палладия(II).
. Этот эффект, судя по кинетике
процесса есть ТИЭ, появление которого связано с разницей констант автопротолиза
Kw D2O и H2O (Kw H2O в 5.13 раз выше Kw D2O), и, соответственно, констант
кислотной диссоциации аквакомплексов палладия(II).
Пример. При изучении кинетики реакций окислительного карбонилирования ацетилена в растворах комплексов Pd(I) с образованием ангидридов малеиновой (МА) и янтарной (ЯА) кислот, акриловой (АК) и пропионовой (ПК) кислот установлено, что МА и ЯА образуются с КИЭ ~ 1.
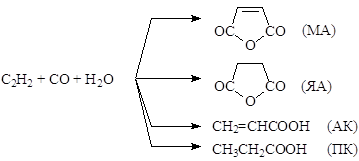
Этот факт свидетельствует о том, что в образовании МА молекула H2O разрывается за необратимой медленной стадией. ЯА образуется из МА с переносом атома водорода от PdH, но также за медленной стадией. КИЭ образования АК и ПК составляют 1.8 и 2.5, соответственно, что говорит о разрыве Pd-H (и образовании С-Н) в медленной стадии. Образование МА и ЯА сопровождается сильным обменом С-Н/C-D, что дает дополнительную информацию о механизме процесса.
Пример. При изучении механизма реакции
![]()
предложили 41 гипотезу о механизме реакции. На
основании измерения КИЭ при переходе от CH3OH к CH3OD были отброшены 32
гипотезы (![]() ).
Еще четыре гипотезы были отклонены на основании предварительных экспериментов.
Оставшиеся пять гипотез являются работающими гипотезами до настоящего времени.
).
Еще четыре гипотезы были отклонены на основании предварительных экспериментов.
Оставшиеся пять гипотез являются работающими гипотезами до настоящего времени.
Анализ селективности процесса
Селективность процесса характеризует долю исходного ключевого реагента, превратившегося в целевой продукт, от общего количества ключевого реагента, превратившегося по всем маршрутам.
Интегральная селективность процесса образования вещества i по ключевому реагенту k
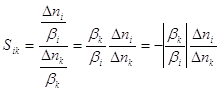 , (1)
, (1)
Поскольку ![]() , получим
, получим
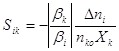 , (2)
, (2)
где Xk – степень конверсии k-того реагента.
Дифференциальная селективность sik определяется уравнением (3)
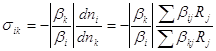 , (3)
, (3)
где Rj – скорость реакции j, bij – стехиометрические коэффициенты i-того продукта в j-той реакции, bkj – тоже для k-того исходного реагента в j-той реакции. Величина sik > 0, так как bkj < 0. В стационарном реакторе полного смешения sik = Sij.
Анализ изменений селективности от концентраций реагентов (или от степени превращения Xk) полезен для предварительной дискриминации гипотез.
Пример. Рассмотрим две параллельные реакции и скорости R1 и R2 по итоговым уравнениям двух маршрутов в проточном реакторе полного смешения:
![]()
![]()
Рассмотрим селективность расходования реагента А по первой реакции
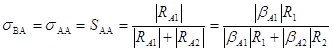 (4)
(4)
(![]() ,
,![]() ,
,![]() ).
).
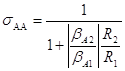 (5)
(5)
Если реакции имеют простую кинетику, например, ![]() и
и ![]() ,
,
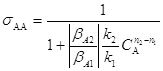 (6)
(6)
Поскольку ![]() , то из уравнения (6) следует, что
sАА
будет не зависеть от XA при n2 = n1, расти с увеличением XA при n2 > n1 и
падать с ростом XA при n2 < n1.
, то из уравнения (6) следует, что
sАА
будет не зависеть от XA при n2 = n1, расти с увеличением XA при n2 > n1 и
падать с ростом XA при n2 < n1.
Как видно из уравнений (5) и (6) при анализе
зависимостей sik от Ck (XA) мы имеем дело с изменением отношения скоростей
реакций (или сумм отношений скоростей в более сложных случаях). Отношения
скоростей в многомаршрутных процессах с линейными механизмами в стационарных и
квазистационарных условиях всегда существенно проще, чем выражения для Rp,
поскольку многочленные полиномы, стоящие в знаменателе кинетических уравнений (![]() ) при этом
сокращаются. Рассмотрим этот вопрос подробнее.
) при этом
сокращаются. Рассмотрим этот вопрос подробнее.
Анализ узлов сопряжения
Все интермедиаты в реакционной сети, которые превращаются по двум и более стадиям, не считая обратной стадии образования интермедиата, образуют так называемые узлы сопряжения. Простейший узел – параллельно-последовательная реакция
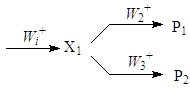
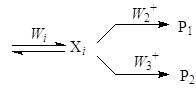
Более сложный узел включает дополнительные интермедиаты:
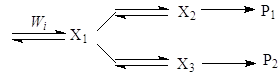
Во всех случаях отношение R1/R2 намного более информативно для целей дискриминации, чем сами скорости R1 и R2.
Рассмотрим узел сопряжения (7)
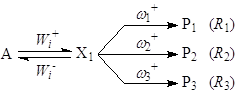 , (7)
, (7)
где wj – веса стадий j.
В стационарных условиях
![]()
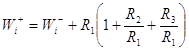
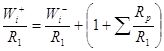
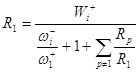 (8)
(8)
В уравнение (8) входит скорость образования X1,
отношение весов ![]() при постоянных концентрациях,
входящих в
при постоянных концентрациях,
входящих в ![]() и
переменная сумма
и
переменная сумма ![]() , которая меняется в зависимости
от
, которая меняется в зависимости
от ![]() и
и ![]() . Выполнимость
уравнения (8)
. Выполнимость
уравнения (8)
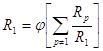
может служить подтверждением схемы (7) и
позволяет найти ![]() и
и ![]() .
.
Другой вариант анализа схемы (7):
![]() (9)
(9)
Поскольку ![]()
![]() . Тогда
. Тогда
![]() , откуда
, откуда
![]() или (10)
или (10)
![]() (11)
(11)
(при сохранении концентраций, входящих в ![]() и
и ![]() постоянными).
постоянными).
Пример. Изучали реакцию
![]() (12)
(12)
в системе CuCl (3 – 9![]() ) – NH4Cl – HCl – H2O в условиях
высокого катионного фона ([NH4+] = 12
) – NH4Cl – HCl – H2O в условиях
высокого катионного фона ([NH4+] = 12![]() ) в безградиентом проточном
реакторе при поддержании постоянными активностей Cu+ и Cl– и концентраций
каталитически активных комплексов CumCln(n – m)–. При добавлении в этот раствор
CuCl2 (5·10–3 – 170·10–3 М) скорость образования винилхлорида RВХ понижается, и
появляются два продукта окислительного хлорирования ацетилена –
1,2-дихлорэтилен (ДХЭ) и винилиденхлорид (ВДХ):
) в безградиентом проточном
реакторе при поддержании постоянными активностей Cu+ и Cl– и концентраций
каталитически активных комплексов CumCln(n – m)–. При добавлении в этот раствор
CuCl2 (5·10–3 – 170·10–3 М) скорость образования винилхлорида RВХ понижается, и
появляются два продукта окислительного хлорирования ацетилена –
1,2-дихлорэтилен (ДХЭ) и винилиденхлорид (ВДХ):
![]() (13)
(13)
![]() , (14)
, (14)
скорости образования которых RДХЭ и RВДХ проходят через максимум по мере увеличения [CuCl2]. При малых [CuCl2] наблюдается слабый рост ВХ от [CuCl2]. Для создания стационарного режима по [CuCl2] концентрацию Cu(II) поддерживали постоянной, окисляя Cu(I) электрохимически в ходе реакции. Скорость синтеза ВХ до добавления CuCl2 описывается простым уравнением
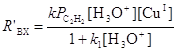 (15)
(15)
в соответствии со схемой механизма
![]() (16)
(16)
Падение RВХ при добавлении CuCl2 говорит о взаимной связи процессов (12), (13) и (14) и о наличии узла сопряжения.
Первая гипотеза о простом узле сопряжения стадий образования ВХ и ДХЭ позволяет рассмотреть отношение RДХЭ и RВХ.
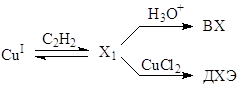
Зависимость отношения RДХЭ/RВХ от [CuCl2] при постоянных остальных параметрах системы описывается уравнением (16)
![]() . (16)
. (16)
Второй вариант узла сопряжения связывает ВДХ и ВХ.
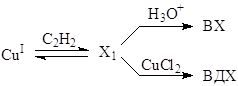
Отношение RВДХ/RВХ = f [CuCl2] описывается более сложной зависимостью, простейший вид которой подчиняется уравнению (17)
![]() . (17)
. (17)
Квадрат по концентрации CuCl2 в числителе уравнений (16) и (17) свидетельствует о последовательном участии двух молекул CuCl2 в стадиях окисления Х1 до ДХЭ и ВДХ
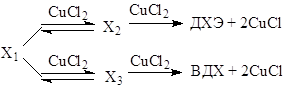 (18)
(18)
При этом стадия образования Х2 квазиравновесна (16), а стадия образования Х3 – нет (17). Чтобы найти выражение для RВХ, RДХЭ и RВДХ не хватает еще одного уравнения. Воспользуемся зависимостью (11). Эксперимент показал, что выполняется линейное соотношение (11) в форме уравнения (19) при варьировании [CuCl2]
![]() (19)
(19)
Решение системы (16), (17) и (19) дает уравнения (при [H3O+] = const)
![]() (20)
(20)
![]() (21)
(21)
![]() (22)
(22)
![]() (23)
(23)
Уравнения (20 – 23) описывают экспериментальные зависимости Rp = f([CuCl2]) и соотношение скоростей Rp.
Исследования различных модельных реакций и отклонение других гипотез образования ВДХ, например, через ClCºCH
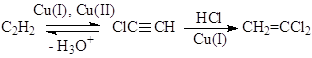
позволило предложить механизм с участием s-металлоорганических соединений Cu(I) и Cu(II)
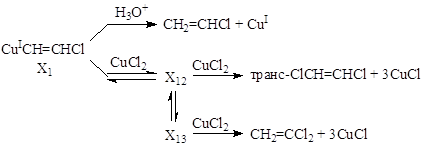
Образование X12 квазиравновесно. Предположительно, X12 – комплекс [CuIICl2·CuICH=CHCl « ClCuI·ClCuIICH=CHCl]. Реакция X12 c CuCl2 дает CuIICl2·ClIICuCH=CHCl, быстро распадающийся до ClCH=CHCl и 3CuCl. Превращение X12 « X13 с последующим действием второй молекулы CuCl2 приводит к ВДХ. Например, образование h2-винильного комплекса из X12 с последующим превращением его в карбеновый комплекс, распадающийся до CuI и CH2=CCl2
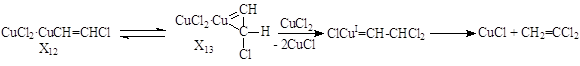
Рассмотренный пример показывает, насколько полезно создание искусственных разветвлений, искусственных узлов сопряжения при изучении каталитических реакций, например, реакции (12). Введение CuCl2 позволило получить дополнительную информацию о природе Х1.
На завершающем этапе выбора модели решается обратная задача химической кинетики.