Реферат: Жидкофазное и газофазное гидрирование углеводородов
Раздел: Рефераты по химии
Тип: реферат
Особенности оформления реакционного узла жидкофазного гидрирования углеводородов. Классификация реакций жидкофазного гидрирования в зависимости от формы применяемого катализатора. Влияние термодинамических факторов на выбор условий процесса. Селективность реакций гидрирования.
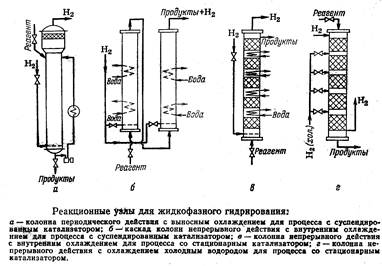
Жидкофазное гидрирование проводят путем барботирования водорода через жидкую реакционную массу. Таким способом всегда гидрируют высококипящие вещества (жиры, высшие карбоновые кислоты и их эфиры, динитросоединения), поскольку для перевода их в состояние насыщенного пара потребовались бы слишком большие затраты. Однако в жидкой фазе можно гидрировать и более летучие соединения (при высоком давлении).
Процессы жидкофазного гидрирования можно классифицировать в зависимости от формы применяемого катализатора:
· С гомогенным катализатором, нередко получаемым непосредственно в массе гидрируемого вещества. Такой катализатор очень активен, но его трудно отделять от гидрогенизата при последующей переработке (чаще всего – комплексные металлоорганические соединения).
· с суспендированным в реакционной среде катализатором, который или формируется в реакционной среде, например, при разложении неустойчивых соединений или измельчается вне реактора и вводится в виде пасты в сырье. В обоих случаях достаточно сложным оказывается отделение катализатора от продуктов реакции; кроме того, возникают затруднения, связанные с эрозией аппаратов, трубопроводов, арматуры. Катализатор быстро истирается, поэтому суспендирование применяется для быстро дезактивирующихся катализаторов.
· Со стационарным (неподвижным) гранулированным катализатором. Гранулы должны быть достаточно крупными, чтобы их не выносили из реактора потоки продуктов реакции и водородсодержащие газы. Исключается стадия фильтрования гидрогенизата. Т.к. перегрузка катализатора – сложная операция, а простои во время перегрузки снижают технико-экономические показатели процесса, то стационарный катализатор следует применять в случае его высокой стабильности (продолжительного срока службы).
Многие процессы гидрирования протекают через ряд промежуточных стадий. Так, карбоновые кислоты, альдегиды и кетоны восстанавливаются последовательно в спирты и углеводороды:
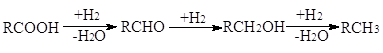
а нитрилы – в имины, амины и углеводороды:
![]()
При дальнейшем развитии этих реакций может произойти гидрогенолиз органического соединения с образованием нежелательных продуктов деструкции
![]()
Во всех случаях всегда требуется остановить реакцию на определенной стадии, т.е. провести частичное гидрирование исходного вещества, ограничив протекание последующих превращений. С другой стороны, в молекуле органического соединения нередко находятся несколько функциональных групп, способных к гидрированию, причем должно протекать гидрирование только одной из них. Так, при гидрировании ненасыщенных кислот можно получить ненасыщенный спирт или ненасыщенную кислоту, из фенолов – спирт циклогексанового ряда или ароматический углеводород.
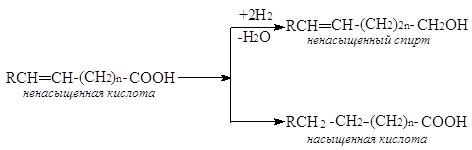 |
|||
На одном и том же катализаторе селективность процесса зависит от ряда факторов, в том числе от относительной реакционной способности органических веществ или отдельных функциональных групп и от их способности адсорбироваться поверхностью катализатора (например, двойные связи арилолефинов гидрируются быстрее ароматического ядра; альдегидные группы быстрее кетонных). Но иногда хемосорбция и реакционная способность изменяются в противоположных направлениях. Тогда вещество лучше сорбируемое вытесняет с поверхности катализатора другой реагент или промежуточный продукт и гидрируется в первую очередь. Этим объясняется то, что ацетилен и его гомологи можно селективно гидрировать в соответствующие олефин6ы, несмотря на более высокую реакционную способность образующихся олефинов.
Сорбционная способность катализатора по отношению к различным веществам или функциональным группам является важным показателем, учет которого при выборе контакта служит средством повышения селективности реакции. Металлические катализаторы (особенно платина, палладий, никель) не имеют специфической способности к адсорбции полярных соединений и функциональных групп, и на поверхности их легче протекает адсорбция реагентов по С-С связи. Поэтому ненасыщенные кетоны, карбоновые кислоты и некоторые производные ароматических углеводородов гидрируются на металлических контактах главным образом по С-С связям с сохранением полярной группы.
Оксидные катализаторы, имеющие полярную кристаллическую решетку, обладают специфической сорбционной способностью к полярным группам органических соединений. Полифункциональное соединение при адсорбции на поверхности оксидного катализатора оказывается ориентированным по полярной группе, в связи с чем, ненасыщенные и ароматические альдегиды, кетоны, карбоновые кислоты и другие вещества гидрируются на оксидных катализаторах преимущественно по кислородсодержащим группам с сохранением ненасыщенных связей.
Независимо от выбора катализатора и других условий на селективности гидрирования сильно влияет температура. Обычно, чем ниже температура, тем селективнее можно провести процесс по более реакционно-способным группам или остановить его на определенной промежуточной стадии. Повышение температуры, соответственно, способствует более глубоким превращениям. Существенно, что нежелательные побочные реакции (гидрогенолиз, крекинг, дегидроциклизация и т.д.) имеют более высокую энергию активации, чем гидрирование. Так, для крекинга н-бутана Еакт. = 280 кДж/моль, а для его гидрирования »180-190 кДж/моль, что позволяет повысить селективность путем понижения температуры. Поскольку при уменьшении температуры снижается скорость процесса и уменьшается производительность реактора, то необходимо для каждого конкретного случая найти область температур, соответствующую минимуму энергетических и экономических затрат и максимуму селективности. Ограничения на выбор этого оптимума налагает обратимость реакции гидрирования.
При прочих равных условиях селективность зависит от времени контакта, определяющего фактическую степень конверсии исходного вещества. Чем ближе она к равновесной, тем значительнее развитие последовательных реакций более глубокого гидрирования. Обычно гидрирование проводят до высокой (до 90%) степени конверсии. Но для каждого процесса время контакта определяется экспериментально.
Все реакции присоединения водорода являются экзотермическими и обратимыми. Теплота гидрирования алкенов с двойной связью на конце молекулы (пропилен, бутен-1) больше, чем алкенов с двойной связью, расположенной ближе к середине цепи (бутен-2).
При расчете на одну молекулу присоединяющегося водорода тепловой эффект наиболее высок для соединений со связью СºС.
СНºСН + 2Н2 ® СН3-СН3 (-DН = 311 кДж/моль)
Для ароматических систем он меньше, чем для олефинов, что может быть обусловлено нарушением устойчивости ароматической системы.
С6Н6 + 3Н2 ® С6Н12 (-DН = 206 кДж/моль)
RCH=CH2 + H2 ® RCH2-CH3 (-DН = 113-134 кДж/моль)
При гидрировании двойной связи между углеродом и кислородом в карбонильных соединениях тепловой эффект ниже, чем для двойной углерод - углеродной связи
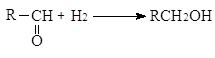
(-DН = 67-68 кДж/моль)
при этом гидрирование альдегидов более экзотермично, чем гидрирование кетонов:
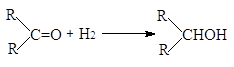
(-DН = 67-68 кДж/моль)
Близкий к ним тепловой эффект (в расчете на одну молекулу водорода) имеет гидрирование нитрилов:
RCN + 2H2 ® RCH2NH2 (-DН = 134-159 кДж/моль).
Наименьший тепловой эффект наблюдается при гидрировании кислот:
RCOOH + 2H2 ® RCH2OH + H2O (-DН = 38-42 кДж/моль)
и деструктивном гидрировании связи С-С
-СН2-СН2- + Н2 ® -СН3 + -СН3 (-DН = 42-63 кДж/моль)
Особенности оформления реакционного узла газофазного гидрирования углеводородов. Равновесие реакций гидрирования. Специфика применения катализаторов различного типа при проведении реакций гидрирования. Кинетика реакций гидрирования.
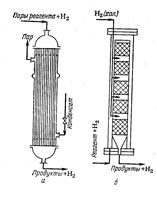
Реакционные аппараты для газофазного гидрирования:
а) трубчатый реактор; б) колонна со сплошными слоями гетерогенного катализатора и охлаждением холодным водородом
Реакция гидрирования обратима. Обратный процесс – дегидрирование. Вследствие экзотермичности реакций гидрирования равновесная степень превращения увеличивается при понижении температуры.
Термодинамически наиболее благоприятно протекает гидрирование ацетиленовых производных, наименее – гидрирование кислот.
Поскольку при гидрировании (за исключением деструктивного) всегда происходит уменьшение объема. Для увеличения равновесной степени превращения, особенно при высокой температуре, применяют повышенное давление. Другим методом повышения степени превращения является применение избытка водорода по сравнению со стехиометрическим от 5010-кратного до 100-кратного и более. Часто одновременно применяется и повышенное давление, и избыток водорода.
Равновесие благоприятно для гидрирования неразветвленных углеводородов с небольшой молекулярной массой, а также для гидрирования низших олефинов, диенов и, как уже говорилось выше, для ацетиленовых углеводородов, причем наличие фенильных заместителей и разветвления углеродной цепи сказывается отрицательно. Менее выгодны условия гидрирования альдегидов, нитрилов, кетонов и ароматических ядер. Если провести сравнение для температуры, при которой Кр = 1, получим следующий ряд способности к гидрированию, учитывающий только термодинамические факторы:
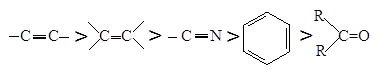
В процессах гидрирования, сопровождающихся выделением воды, равновесие обычно смещено вправо в большей мере, чем в других случаях.
Для гидрирования вещества Кр равна А + Н2 ® АН2
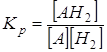 .
.
Для реакций перераспределения водорода А + ВН2 ® АН2 + В

Равновесие реакции перераспределения будет смещено вправо, когда в термодинамическом отношении вещество А более склонно к гидрированию, чем вещество В, например:
3RCH=CH2 + C6H12 ® 3RCH2-CH3 + C6H6
Гидрирование может протекать в гомогенной (газовой или жидкостной), гетерогенной (газ-жидкость, газтвердое тело, жидкостьтвердое тело) системах в присутствии катализаторов или без них.
Скорость реакций гидрирования в общем случае может зависеть от диффузионных и кинетических факторов. Первые из них играют тем меньшую роль, чем интенсивнее перемешивание и чем ниже температура. Скорость гидрирования определяется в большей степени, чем для других процессов, влиянием следующих факторов:
· величиной окислительно-восстановительного потенциала системы;
· скоростью диффузии реагентов из одной фазы в другую;
· скоростями адсорбции, хемосорбции и диффузии в адсорбированный слой;
· ориентацией адсорбированных молекул и т.д.
Процессы гидрирования обычно осуществляются в условиях, когда равновесие реакции значительно смещено вправо и можно пренебречь обратной реакцией дегидрирования. Кроме того, насыщенный продукт гидрирования имеет небольшой адсорбционный коэффициент и поэтому обычно не входит в кинетическое уравнение процесса. И, наоборот, при высоком давлении становится существенной сорбция не только исходного вещества, но и водорода.
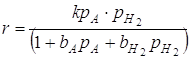 ,
,
где Р – парциальные давления реагентов;
b – адсорбционные коэффициенты реагентов.
С такими катализаторами, как платина, палладий, никель, энергично сорбирующими водород, скорость реакции при умеренных температурах (100оС) не зависит от парциального давления водорода. Оно начинает влиять на скорость только при повышении температуры - вначале незначительно, а затем пропорционально возрастанию давления. Наблюдается и отчетливое самоторможение реакции исходными ненасыщенными соединениями. Как уже говорилось, на оксидных катализаторах сорбция водорода менее значительна, чем на металлах, вследствие чего скорость обычно зависит от парциального давления водорода линейно. Этим обусловлена большая эффективность применения высоких давлений и избытка водорода при гидрировании на оксидных катализаторах.
В жидкофазных процессах высокое давление оказывает дополнительное влияние, повышая растворимость водорода в реакционной массе. Возможна линейная, квадратичная и даже более сильная зависимость скорости реакции от давления. Так, при гидрировании этиллаурата в лауриловый спирт на медь – хромоксидном катализаторе скорость возрастает в 7 раз при повышении давления от 10 до 20 МПа, а с увеличением до 30 Мпа – в 28 раз.
Влияние температуры. При повышении температуры на 30-50оС скорость примерно удваивается, что соответствует энергии активации »21-42 кДж/моль.
По способности к гидрированию разные классы соединений располагаются в следующие ряды:
· Олефин > ацетилен и его производные > ароматические углеводороды;
· Этилен > пропилен > бутен-2 > i-бутилен
138 11 1,3 1 Относительная скорость гидрирования
Т.е. скорость уменьшается с увеличением количества и степени разветвления заместителей. Скорость зависит также и от природы катализатора (Pt > Pd > Ni).
Соотношение между скоростями, установленное для чистых углеводородов не сохраняется при гидрировании их смесей. Поэтому, несмотря на то, что скорость гидрирования чистого бутадиена в бутен и чистого бутена в бутан практически являются теми же, в смеси этих соединений (бутадиен – бутен - бутан) гидрирование бутадиена в бутен протекает намного быстрее. Возможно, это объясняется большей величиной коэффициента хемосорбции бутадиена, возможно, каким-либо возникающим в данной системе эффектом синергизма.
На одном и том же катализаторе селективность процесса зависит от ряда факторов, в том числе от относительной реакционной способности органических веществ или отдельных функциональных групп и от их способности адсорбироваться поверхностью катализатора (например, двойные связи арилолефинов гидрируются быстрее ароматического ядра; альдегидные группы быстрее кетонных). Но иногда хемосорбция и реакционная способность изменяются в противоположных направлениях. Тогда вещество лучше сорбируемое вытесняет с поверхности катализатора другой реагент или промежуточный продукт и гидрируется в первую очередь. Этим объясняется то, что ацетилен и его гомологи можно селективно гидрировать в соответствующие олефин6ы, несмотря на более высокую реакционную способность образующихся олефинов.
Сорбционная способность катализатора по отношению к различным веществам или функциональным группам является важным показателем, учет которого при выборе контакта служит средством повышения селективности реакции. Металлические катализаторы (особенно платина, палладий, никель) не имеют специфической способности к адсорбции полярных соединений и функциональных групп, и на поверхности их легче протекает адсорбция реагентов по С-С связи. Поэтому ненасыщенные кетоны, карбоновые кислоты и некоторые производные ароматических углеводородов гидрируются на металлических контактах главным образом по С-С связям с сохранением полярной группы.
Оксидные катализаторы, имеющие полярную кристаллическую решетку, Обладают специфической сорбционной способностью к полярным группам органических соединений. Полифункциональное соединение при адсорбции на поверхности оксидного катализатора оказывается ориентированным по полярной группе, в связи, с чем ненасыщенные и ароматические альдегиды, кетоны, карбоновые кислоты и другие вещества гидрируются на оксидных катализаторах преимущественно по кислородсодержащим группам с сохранением ненасыщенных связей.