Реферат: Ионные реакции в растворах. Солевой эффект (в ТАК)
Ионные реакции в растворах. Солевой эффект (в ТАК)
Теория активированного комплекса позволяет элегантно объяснить специфические особенности кинетики ионных реакций в жидкой фазе.
Теория Активированного Комплекса – Теория Переходного Состояния - Теория Абсолютных Скоростей химических реакций... Всё это наименования одной и той же теории, в которую ещё в 30-е годы оформились попытки представить процесс активации с помощью и достаточно детальных, и вместе с тем всё же достаточно общих, моделей, построенных на базе статистической механики и квантовой химии (квантовой механики), комбинируя их и создавая иллюзию индивидуального анализа конкретного химического превращения уже на стадии перестройки электронно-ядерной структуры реагентов.
Сама задача кажется очень сложной, и поэтому в ТАК неизбежно образовалось довольно много логических неясностей... Всё же это наиболее общая и плодотворная из теоретических концепций, посредством которых в настоящее время описывают элементарные процессы, и её возможности не ограничены рамками лишь химического элементарного акта. С нею оказалось тесно связано развитие современной химической кинетики. К ней привязаны новейшие алгоритмы и графические приёмы компьютерной химии, и на её основе быстро развивается орбитальная теория химической реакционной способности...
И это далеко не всё! На основе ТАК оказалось возможно единообразно проанализировать множество физико-химических явлений и многих макроскопических свойств веществ, что, на первый взгляд, выглядят уделом лишь научной эмпирики, казалось бы безнадёжно недоступной для теоретического осмысления. Ряд таких ситуаций читатель найдёт в великолепной, хотя и давней, книге Глесстона, Эйринга и Лейдлера “Теория абсолютных скоростей”, написанной творцами этой теории...
Рассмотрим элементарные положения теории активированного комплекса, включая:
- кинетическую схему активации через промежуточное переходное состояние,
- квазитермодинамику активации через образование активированного комплекса,
- размерность константы скорости реакции второго порядка в ТАК.
Простейшая кинетическая модель активации в ТАК:
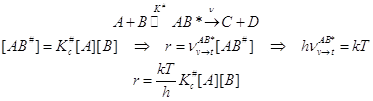 (6.1)
(6.1)
Первая стадия механизма активации бимолекулярная. Она обратимая, на ней образуется активированный комплекс, а он далее распадается по двум маршрутам: а) обратно в реагенты, с которыми он находится в равновесии, и для этого процесса следует ввести константу равновесия, б) в продукты реакции и этот финальный процесс характеризуется некоторой механической частотой распада. Сочетая эти стадии, несложно рассчитать константу скорости реакции. Удобно рассматривать превращение в газовой фазе.
Константа равновесия обратимой стадии может быть выражена следующим способом.
Если стандартные состояния в газовой фазе выбраны согласно обычному термодинамическому правилу, и стандартизованы парциальные давления газообразных участников реакции, то это означает:
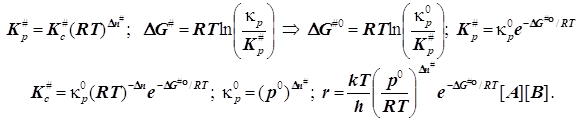
Внимание! Отсюда следует выражение для константы скорости бимолекулярной реакции в ТАК, не вызывающее сомнений в размерности констант скоростей бимолекулярных реакций:
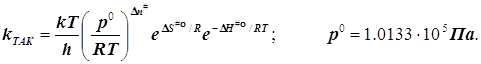 (6.2)
(6.2)
В учебниках чаще всего приводится не столь прозрачное выражение, построенное на иной стандартизации состояний - стандартизуют концентрацию, и в итоге возникает размерность константы скорости, внешне соответствующая моно-, а не би молекулярной реакции. Размерности концентраций оказываются как бы скрыты. У Эйринга, Глесстона и Лейдлера - самих творцов ТАК в книге «Теория абсолютных скоростей реакций» есть анализ, где учтена стандартизация состояний по давлениям. Если стандартным считать состояние с единичными концентрациями реагентов и продуктов, то формулы слегка упростятся, а именно:
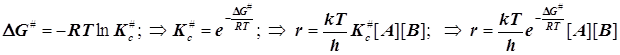 Отсюда следует обычно представленное в
учебниках выражение для константы скорости согласно ТАК:
Отсюда следует обычно представленное в
учебниках выражение для константы скорости согласно ТАК: ![]() (6.3)
(6.3)
Если не выделить роль стандартного состояния, то теоретическая константа скорости бимолекулярного превращения может обрести чужую размерность, обратную времени, которая будет отвечать мономолекулярной стадии распада активированного комплекса. Активационные величины S#0 и H#0 нельзя считать обычными термодинамическими функциями состояния. Они не сопоставимы с обычными характеристиками пробега реакции уже потому, что методов их прямого термохимического измерения просто не существует... По этой причине их можно назвать квазитермодинамическими характеристиками процесса активации.
При образовании частицы
активированного комплекса из двух исходных частиц имеет место ![]() , и в результате получается
, и в результате получается
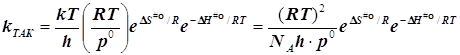 (6.4)
(6.4)
Размерность константы скорости обычная для реакции второго порядка:
![]()
Эмпирическая энергия активации по Аррениусу и её сравнение с близкими аналогичными активационными параметрами (энергиями) ТАС и ТАК:
Основа - уравнение Аррениуса в
дифференциальной форме: ![]()
1) в ТАС получаем:
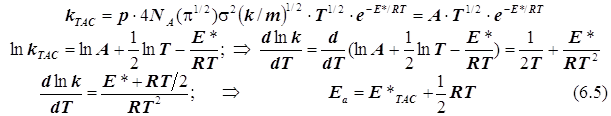
2.1) ТАК. Случай 1. (Общий подход при условии стандартизации концентраций)
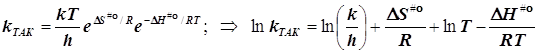
подстановка в уравнение Аррениуса даёт
![]()
2.2) ТАК.
Случай 2. (Частный случай бимолекулярной стадии активации ![]() )
)
Энергия активации по Аррениусу для бимолекулярной реакции:
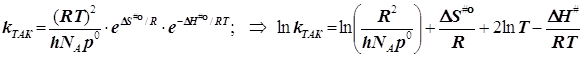
Внимание!!! Полагаем чаще всего ![]()
2.2) Исходя из стандартизации давления, получаем энергию активации:
![]()
![]()
![]() (6.7)
(6.7)
2.3) Это же получается для бимолекулярной реакции и при стандартизации концентрации:
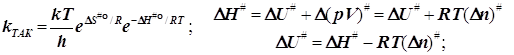 (6.8)
(6.8)
![]()
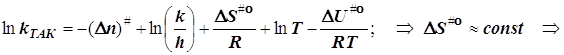
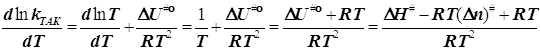
![]()
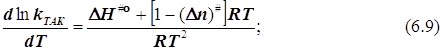
в бимолекулярном акте активации n#= -1, и![]() (6.10)
(6.10)
Результат: Формула, связывающая энергию активации Аррениуса с квазитермодинамическими функциями активации теории переходного состояния, не зависит от выбора стандартного состояния.
3. Адиабатические потенциалы и потенциальные поверхности
Пример. Реакция обмена одного из атомов в молекуле водорода на дейтерий
(Это простейший из любых возможных примеров)
![]()
По мере сближения атома дейтерия с молекулой водорода
наблюдается разрыхление старой двухцентровой химической связи H-H и постепенное оформление новой связи H-D, так что энергетическая модель реакции дейтерообмена в
молекуле водорода может быть построена как постепенное перемещение исходной
трёхатомной системы к конечной согласно схеме: 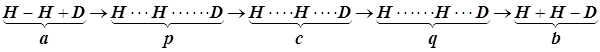
К ним могут относиться превращения разной природы: и окисление-восстановление, и обмен лигандами между комплексными ионами, и их объединяет кинетическая схема вида:
![]() .
.
Первая стадия равновесная, а за нею следует медленное
превращение в продукты. Константа скорости в ТАК, равна ![]() . (10.1)
. (10.1)
Она была построена для смеси идеальных газов и согласно принципу детального равновесия содержит константу равновесия Kc# первой стадии образования активированного комплекса. Через неё в активационное соотношение вводятся концентрации реагентов, но квазитермодинамические активационные функции H# и S# для неидеальной смеси реагентов полагается связать уже не с Kc#, а с Ka#. При переходе к неидеальным системам необходимо скорректировать полученное в ТАК выражение константы скорости:
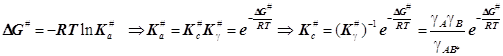 (10.2)
(10.2)
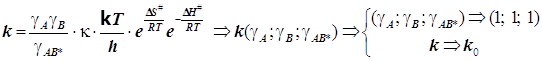
Искомая коррекция выражается уравнением Брёнстеда-Бьеррума в виде:
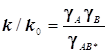 или в
логарифмической форме
или в
логарифмической форме 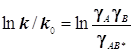 . (10.3)
. (10.3)
С его помощью были исследованы некоторые реакции обмена лигандами между комплексными ионами переходных металлов (d-элементов). Скорости этих реакции обычно сравнительно невелики и доступны для традиционных классических методов измерений. Обратимся к теории растворов сильных электролитов по Дебаю- Хюккелю.
Для миллимолярных (почти «предельно разбавленных») растворов справедливо:
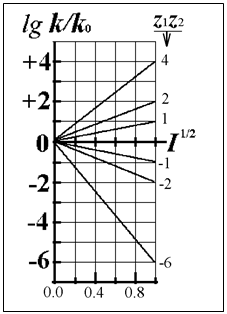
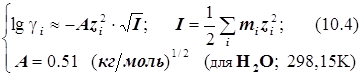
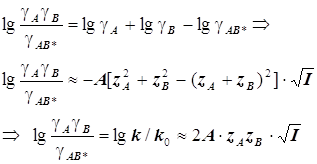
![]()
Константы скоростей согласно формуле (8.5) линейно зависят от квадратного корня из ионной силы раствора. Графики этих зависимостей (точнее, касательные к ним) образуют пучок прямых, и значения их угловых коэффициентов оказываются дискретными - «квантованными», поскольку заряды ионов zi и их произведения zAzB принадлежат к ряду целых (и положительных, и отрицательных) чисел. (рис.21.) Это кинетическое явление называется первичным солевым эффектом.

Реакции в растворах (дополнения)
Особенно интересны реакции, которые можно изучить и в газовой фазе, и в растворе. Их немного:
1) Реакция разложения дийодэтана ![]() описывается единым уравнением Аррениуса как в
газовой фазе, так и в растворе в
описывается единым уравнением Аррениуса как в
газовой фазе, так и в растворе в ![]() . Этот случай является одним из наиболее прямых свидетельств
в пользу единой кинетической модели реакции в газе и в жидкости. В растворе
распределение Максвелла-Больцмана оказывается даже более устойчивым, чем в
газе, и теория активных соударений оказывается весьма корректной.
. Этот случай является одним из наиболее прямых свидетельств
в пользу единой кинетической модели реакции в газе и в жидкости. В растворе
распределение Максвелла-Больцмана оказывается даже более устойчивым, чем в
газе, и теория активных соударений оказывается весьма корректной.
2) В обеих фазах изучены также реакции
2.1) ![]()
![]()
(реакция изучена и в газе, и в различных растворах).
2.2) ![]() Механизм
сложный, стадий две. Обе второго порядка. Энергии активации стадий равны.
Скорости мало зависят от растворителя, но зависят от начальной концентрации и
почти совпадают (см. книгу Ерёмина, стр.297). См. медленные реакции в растворе.
Механизм
сложный, стадий две. Обе второго порядка. Энергии активации стадий равны.
Скорости мало зависят от растворителя, но зависят от начальной концентрации и
почти совпадают (см. книгу Ерёмина, стр.297). См. медленные реакции в растворе.
3) Реакция Меншуткина (это медленная реакция)
![]()
Особенность состоит в том, что энергия активации сравнительно мала (всего около 11-13 ккал/пробег реакции) Причина - чрезвычайно малый предэкспонент: 0.028>A, л/моль-1с-1>0.00027. Нормальный предэкспонент равен 2.8*1011 л/моль*с. Он же реально на 8-10 порядков меньше. Стерический фактор лежит на интервале 1.6*10-8>p>5.3*10-10 . Выяснено, что и растворитель здесь не при чём, поскольку в растворе -(в жидкой ароматике) реакция протекает всего в 5 раз медленнее, чем в газовой фазе.
В раздел «Кинетика реакций в конденсированных фазах» обязательно следует включить:
- Влияние жидкой среды на кинетику реакций. Полярная среда. Уравнение Кирквуда.
- Компенсационный эффект. Типовые механизмы и реакционные серии. Гомологические ряды реагентов.
- Особенно важны специфические химические реакции и специфические эффекты в жидкой фазе, как-то: - Эффект клетки (Эффект Франка-Рабиновича).
- Быстрые реакции (рапидные реакции) в растворах (Исследования М. Эйгена).
- Релаксационные методы кинетики.
- Колебательные реакции (Реакция Белоусова).
- Топохимические процессы в твёрдой фазе. Нитроксильные свободные радикалы и органические ферро-магнетики. (чл.-кор.РАН, проф. А.А.Овчинников, проф.В.Н.Спектор, проф. Э.Г.Розанцев, к.х.н. А.В. Чудинов)... Открыл нитроксильные радикалы к.х.н. Олег Львович Лебедев (НИОПИК, г. Долгопрудный).
- Безактивационный химический элементарный акт при сверхнизких температурах -полимеризация формальдегида вблизи абсолютного нуля. Туннельный эффект в активационых процессах (акад.В.И. Гольданский, акад. Н.С. Ениколопов, проф. И.М. Агаянц, Э.В. Прут и др.).
- Процессы молекулярной самоорганизации (Кинетические исследования М. Эйгена и его школы (Геттингенский унив-т). Диалог Пригожина и Эйгена в Линдау о термодинамической или кинетической природе самоорганизации материи и о происхождении жизни. (Bayern, Lindau, Die Tagung der).