Учебное пособие: Коллоидная химия
ВВЕДЕНИЕ
Предметом физической химии является объяснение химических явлений на основе более общих законов физики. Физическая химия рассматривает две основные группы вопросов:
1. Изучение строения и свойств вещества и составляющих его частиц;
2. Изучение процессов взаимодействия веществ.
В курсе физической химии обычно выделяют несколько разделов.
Строение вещества. В этот раздел входят учение о строении атомов и молекул и учение об агрегатных состояниях вещества. Изучение строение вещества необходимо для выяснения важнейших вопросов об образовании молекул из атомов, о природе химической связи, о строении и взаимодействии молекул. Именно в этой своей части физическая химия очень тесно переплетается со всеми направлениями современной химии, поскольку изучение химических свойств вещества вне связи со строением атомов и молекул на современном уровне невозможно.
Химическая термодинамика изучает энергетические эффекты химических процессов; позволяет определить возможность, направление и глубину протекания химического процесса в конкретных условиях.
Химическая кинетика. В этом разделе физической химии изучается скорость и механизм протекания химических процессов в различных средах при различных условиях.
Учение о растворах рассматривает процессы образования растворов, их внутреннюю структуру и важнейшие свойства, зависимость структуры и свойств от природы компонентов раствора.
Электрохимия изучает особенности свойств растворов электролитов, явления электропроводности, электролиза, коррозии, работу гальванических элементов.
Коллоидная химия изучает поверхностные явления и свойства мелкодисперсных гетерогенных систем.
Все разделы физической химии объединяет единая основа – общие законы природы, которые применимы к любым процессам и любым системам, независимо от их строения.
1 ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Термодинамика – наука о взаимопревращениях различных форм энергии и законах этих превращений. Термодинамика базируется только на экспериментально обнаруженных объективных закономерностях, выраженных в двух основных началах термодинамики.
Термодинамика изучает:
1. Переходы энергии из одной формы в другую, от одной части системы к другой;
2. Энергетические эффекты, сопровождающие различные физические и химические процессы и зависимость их от условий протекания данных процессов;
3. Возможность, направление и пределы самопроизвольного протекания процессов в рассматриваемых условиях.
Необходимо отметить, что классическая термодинамика имеет следующие ограничения:
1. Термодинамика не рассматривает внутреннее строение тел и механизм протекающих в них процессов;
2. Классическая термодинамика изучает только макроскопические системы;
3. В термодинамике отсутствует понятие "время".
1.1 ОСНОВНЫЕ ПОНЯТИЯ ТЕРМОДИНАМИКИ
Термодинамическая система – тело или группа тел, находящихся во взаимодействии, мысленно или реально обособленные от окружающей среды.
Гомогенная система – система, внутри которой нет поверхностей, разделяющих отличающиеся по свойствам части системы (фазы).
Гетерогенная система – система, внутри которой присутствуют поверхности, разделяющие отличающиеся по свойствам части системы.
Фаза – совокупность гомогенных частей гетерогенной системы, одинаковых по физическим и химическим свойствам, отделённая от других частей системы видимыми поверхностями раздела.
Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией.
Закрытая система – система, которая обменивается с окружающей средой энергией, но не обменивается веществом.
Открытая система – система, которая обменивается с окружающей средой и веществом, и энергией.
Совокупность всех физических и химических свойств системы характеризует её термодинамическое состояние. Все величины, характеризующие какое-либо макроскопическое свойство рассматриваемой системы – параметры состояния. Опытным путем установлено, что для однозначной характеристики данной системы необходимо использовать некоторое число параметров, называемых независимыми; все остальные параметры рассматриваются как функции независимых параметров. В качестве независимых параметров состояния обычно выбирают параметры, поддающиеся непосредственному измерению, например температуру, давление, концентрацию и т.д. Всякое изменение термодинамического состояния системы (изменения хотя бы одного параметра состояния) есть термодинамический процесс. Обратимый процесс – процесс, допускающий возможность возвращения системы в исходное состояние без того, чтобы в окружающей среде остались какие-либо изменения.
Равновесный процесс – процесс, при котором система проходит через непрерывный ряд равновесных состояний.
Энергия – мера способности системы совершать работу; общая качественная мера движения и взаимодействия материи. Энергия является неотъемлемым свойством материи. Различают потенциальную энергию, обусловленную положением тела в поле некоторых сил, и кинетическую энергию, обусловленную изменением положения тела в пространстве.
Внутренняя энергия системы – сумма кинетической и потенциальной энергии всех частиц, составляющих систему. Можно также определить внутреннюю энергию системы как её полную энергию за вычетом кинетической и потенциальной энергии системы как целого.
Формы перехода энергии от одной системы к другой могут быть разбиты на две группы. В первую группу входит только одна форма перехода движения путем хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота. Теплота есть форма передачи энергии путём неупорядоченного движения молекул. Во вторую группу включаются различные формы перехода движения, общей чертой которых является перемещение масс, охватывающих очень большие числа молекул (т.е. макроскопических масс), под действием каких-либо сил. Таковы поднятие тел в поле тяготения, переход некоторого количества электричества от большего электростатического потенциала к меньшему, расширение газа, находящегося под давлением и др. Общей мерой передаваемого такими способами движения является работа – форма передачи энергии путём упорядоченного движения частиц.
Теплота и работа характеризуют качественно и количественно две различные формы передачи движения от данной части материального мира к другой. Теплота и работа не могут содержаться в теле. Теплота и работа возникают только тогда, когда возникает процесс, и характеризуют только процесс. В статических условиях теплота и работа не существуют. Различие между теплотой и работой, принимаемое термодинамикой как исходное положение, и противопоставление теплоты работе имеет смысл только для тел, состоящих из множества молекул, т.к. для одной молекулы или для совокупности немногих молекул понятия теплоты и работы теряют смысл. Поэтому термодинамика рассматривает лишь тела, состоящие из большого числа молекул, т.е. так называемые макроскопические системы.
1.2 ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Первое начало термодинамики представляет собой закон сохранения энергии, один из всеобщих законов природы (наряду с законами сохранения импульса, заряда и симметрии):
Энергия неуничтожаема и несотворяема; она может только переходить из одной формы в другую в эквивалентных соотношениях.
Первое начало термодинамики представляет собой постулат – оно не может быть доказано логическим путем или выведено из каких-либо более общих положений. Истинность этого постулата подтверждается тем, что ни одно из его следствий не находится в противоречии с опытом. Приведем еще некоторые формулировки первого начала термодинамики:
Полная энергия изолированной системы постоянна;
Невозможен вечный двигатель первого рода (двигатель, совершающий работу без затраты энергии).
Первое начало термодинамики устанавливает соотношение между теплотой Q, работой А и изменением внутренней энергии системы ΔU:
Изменение внутренней энергии системы равно количеству сообщенной системе теплоты минус количество работы, совершенной системой против внешних сил.
![]() (I.1)
(I.1)
![]() (I.2)
(I.2)
Уравнение (I.1) является математической записью 1-го начала термодинамики для конечного, уравнение (I.2) – для бесконечно малого изменения состояния системы.
Внутренняя энергия является функцией состояния; это означает, что изменение внутренней энергии ΔU не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутренней энергии U2 и U1 в этих состояниях:
![]() (I.3)
(I.3)
Следует отметить, что определить абсолютное значение внутренней энергии системы невозможно; термодинамику интересует лишь изменение внутренней энергии в ходе какого-либо процесса.
Рассмотрим приложение первого начала термодинамики для определения работы, совершаемой системой при различных термодинамических процессах (мы будем рассматривать простейший случай – работу расширения идеального газа).
Изохорный процесс (V = const; ΔV = 0).
Поскольку работа расширения равна произведению давления и изменения объема, для изохорного процесса получаем:
![]() (I.1)
(I.1)
![]() (I.4)
(I.4)
![]() (I.5)
(I.5)
Изотермический процесс (Т = const).
Из уравнения состояния одного моля идеального газа получаем:
![]() (I.6)
(I.6)
Отсюда:
![]() (I.7)
(I.7)
Проинтегрировав выражение (I.6) от V1 до V2, получим
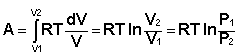 (I.8)
(I.8)
Изобарный процесс (Р = const).
![]() (I.9)
(I.9)
Подставляя полученные выражения для работы различных процессов в уравнение (I.1), для тепловых эффектов этих процессов получим:
![]() (I.10)
(I.10)
![]() (I.11)
(I.11)
![]() (I.12)
(I.12)
В уравнении (I.12) сгруппируем переменные с одинаковыми индексами. Получаем:
![]() (I.13)
(I.13)
Введем новую функцию состояния системы – энтальпию H, тождественно равную сумме внутренней энергии и произведения давления на объем:
![]()
Тогда выражение (I.13) преобразуется к следующему виду:
![]() (I.14)
(I.14)
Т.о., тепловой эффект изобарного процесса равен изменению энтальпии системы.
Адиабатический процесс (Q = 0).
При адиабатическом процессе работа расширения совершается за счёт уменьшения внутренней энергии газа:
![]() (I.15)
(I.15)
В случае если Cv не зависит от температуры (что справедливо для многих реальных газов), работа, произведённая газом при его адиабатическом расширении, прямо пропорциональна разности температур:
![]() (I.16)
(I.16)
1.3 ПРИЛОЖЕНИЯ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
К ХИМИЧЕСКИМ ПРОЦЕССАМ
1.3.1 Закон Гесса
Как известно, большинство химических реакций сопровождаются выделением (экзотермические реакции) либо поглощением (эндотермические реакции) теплоты. Первое начало термодинамики дает возможность рассчитать тепловой эффект химической реакции при различных условиях её проведения.
Тепловой эффект (теплота) химической реакции – количество теплоты, выделившейся либо поглотившейся в ходе реакции. Тепловой эффект относят, как правило, к числу молей прореагировавшего исходного вещества, стехиометрический коэффициент перед которым максимален.
Например, реакцию окисления водорода в химической термодинамике записывают в виде:
Н2 + 1/2 О2 ––> Н2О
и тепловой эффект рассчитывают на 1 моль водорода.
Тепловые эффекты, сопровождающие протекание химических реакций, являются предметом одного из разделов химической термодинамики – термохимии. Определим некоторые понятия термохимии.
Теплота образования вещества – тепловой эффект реакции образования 1 моля сложного вещества из простых. Теплоты образования простых веществ принимаются равными нулю.
Теплота сгорания вещества – тепловой эффект реакции окисления 1 моля вещества в избытке кислорода до высших устойчивых оксидов.
Теплота растворения – тепловой эффект процесса растворения 1 моля вещества в бесконечно большом количестве растворителя. Теплота растворения складывается из двух составляющих: теплоты разрушения кристаллической решетки (для твердого вещества) и теплоты сольватации:
![]()
Поскольку ΔНкр.реш всегда положительно (на разрушение кристаллической решетки необходимо затратить энергию), а ΔНсольв всегда отрицательно, знак ΔНраств определяется соотношением абсолютных величин ΔНкр.реш и ΔНсольв:
![]()
Основным законом термохимии является закон Гесса, являющийся частным случаем первого начала термодинамики:
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Выше было показано, что изменение энтальпии ΔН (тепловой эффект изобарного процесса Qp) и изменение внутренней энергии ΔU (тепловой эффект изохорного процесса Qv) не зависят от пути, по которому система переходит из начального состояния в конечное.
Рассмотрим некоторый обобщенный химический процесс превращения исходных веществ А1, А2, А3 в продукты реакции В1, В2, В3, который может быть осуществлен различными путями в одну или несколько стадий:
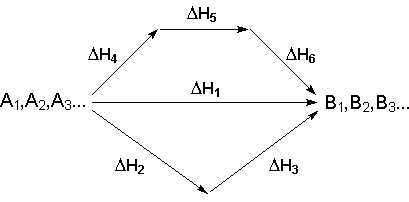
Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:
![]() (I.17)
(I.17)
Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты химических процессов. В термохимических расчетах обычно используют ряд следствий из закона Гесса:
1. Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье – Лапласа).
2. Для двух реакций, имеющих одинаковые исходные, но разные конечные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного конечного состояния в другое.
С + О2 ––> СО + 1/2 О2 ΔН1
С + О2 ––> СО2 ΔН2
СО + 1/2 О2 ––> СО2 ΔН3
![]() (I.18)
(I.18)
3. Для двух реакций, имеющих одинаковые конечные, но разные исходные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного исходного состояния в другое.
С(алмаз) + О2 ––> СО2 ΔН1
С(графит) + О2 ––> СО2 ΔН2
С(алмаз) ––> С(графит) ΔН3
![]() (I.19)
(I.19)
4. Тепловой эффект химической реакции равен разности сумм теплот образования продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты.
![]() (I.20)
(I.20)
5. Тепловой эффект химической реакции равен разности сумм теплот сгорания исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты.
![]() (I.21)
(I.21)
В качестве примера рассмотрим расчет теплового эффекта реакции окисления одного моля глюкозы (теплота образования кислорода по определению равна нулю):
С6Н12О6 + 6 О2 ––> 6 СО2 + 6 Н2О
![]()
Величины тепловых эффектов химических реакций зависят
от условий, в которых проводятся реакции. Поэтому табличные значения теплот
различных процессов принято относить к стандартному состоянию – температуре 298
К и давлению 101325 Па (760 мм. рт. ст.; 1 атм.); величины тепловых эффектов
при данных условиях называют стандартными тепловыми эффектами и обозначают
ΔН°298 и ΔU°298 соответственно.
1.3.2 Зависимость теплового эффекта реакции от температуры. Закон Кирхгофа
В общем случае тепловой эффект химической реакции зависит от температуры и давления, при которых проводится реакция. Влиянием давления на ΔН и ΔU реакции обычно пренебрегают. Влияние температуры на величины тепловых эффектов описывает закон Кирхгофа:
Температурный коэффициент теплового эффекта химической реакции равен изменению теплоемкости системы в ходе реакции.
Продифференцируем ΔН и ΔU по температуре при постоянных давлении и температуре соответственно:
![]() (I.22)
(I.22)
![]() (I.23)
(I.23)
Производные энтальпии и внутренней энергии системы по температуре есть теплоемкости системы в изобарных и изохорных условиях Cp и Cv соответственно:
![]() (I.24)
(I.24)
![]() (I.25)
(I.25)
Подставив выражения (I.24, I.25) в (I.22, I.23), получаем математическую запись закона Кирхгофа:
![]() (I.26)
(I.26)
![]() (I.27)
(I.27)
Для химического процесса изменение теплоемкости задается изменением состава системы и рассчитывается следующим образом:
![]() (I.28)
(I.28)
![]() (I.29)
(I.29)
Если проинтегрировать выражения (I.26, I.27) от Т = Т1 до Т = Т2, считая ΔСp (ΔСv) не зависящим от температуры, получим интегральную форму закона Кирхгофа:
![]() (I.30)
(I.30)
![]() (I.31)
(I.31)
Поскольку обычно известны табличные значения стандартных тепловых эффектов ΔН°298 и ΔU°298, преобразуем выражения (I.30, I.31):
![]() (I.32)
(I.32)
![]() (I.33)
(I.33)
При расчете изменения теплового эффекта реакции в большом интервале температур необходимо учитывать зависимость теплоёмкости от температуры, которая выражается степенным рядом C°p = aT + bT2 + cT3; коэффициенты a, b, c приведены в справочниках.
1.4 ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ. ЭНТРОПИЯ
Первое начало термодинамики утверждает, что при превращении одной формы энергии в другую полная энергия системы не изменяется, однако не указывает никаких ограничений относительно возможности этого процесса. Поэтому первое начало термодинамики позволяет рассчитать энергетический эффект процесса, однако не дает ответа на вопросы о том, будет ли процесс протекать самопроизвольно, о направлении и глубине протекания процесса.
Самопроизвольный процесс – процесс, который может протекать без затраты работы извне, причем в результате может быть получена работа в количестве, пропорциональном произошедшему изменению состояния системы. Самопроизвольный процесс может протекать или обратимо, или необратимо. Хотя определение обратимого процесса уже приводилось, следует подробнее рассмотреть это понятие. Чтобы самопроизвольный процесс протекал обратимо, необходимо приложить извне такое сопротивление, чтобы переход был очень медленным и при бесконечно малом изменении противодействующей силы процесс мог пойти в обратном направлении. В случае обратимо происходящего изменения состояния системы производится максимальное количество работы. Всякий реальный процесс в какой-то степени является необратимым, и получаемая работа меньше максимально возможного теоретического количества.
Вынужденный процесс – процесс, для протекания которого требуется затрата работы извне в количестве, пропорциональном производимому изменению состояния системы.
Второе начало термодинамики дает возможность определить, какой из процессов будет протекать самопроизвольно, какое количество работы может быть при этом получено, каков предел самопроизвольного течения процесса. Далее, второе начало термодинамики дает возможность определить, какими должны быть условия, чтобы нужный процесс протекал в необходимом направлении и в требуемой степени, что особенно важно для решения различных задач прикладного характера. Подобно первому, второе начало термодинамики выведено непосредственно из опыта. В то же время второе начало термодинамики имеет ограниченную область применения: оно применимо лишь к макроскопическим системам. Ниже приведены некоторые формулировки второго начала термодинамики:
Теплота не может самопроизвольно переходить от менее нагретого тела к более нагретому (постулат Клаузиуса).
Невозможен процесс, единственным результатом которого является превращение теплоты в работу.
Невозможно построить машину, все действия которой сводились бы к производству работы за счет охлаждения теплового источника (вечный двигатель второго рода).
Рассмотрим работу тепловой машины, т.е. машины, производящей работу за счет теплоты, поглощаемой от какого-либо тела, называемого нагревателем. Нагреватель с температурой Т1 передает теплоту Q1 рабочему телу, например, идеальному газу, совершающему работу расширения А; чтобы вернуться в исходное состояние, рабочее тело должно передать телу, имеющему более низкую температуру Т2 (холодильнику), некоторое количество теплоты Q2, причем
![]() (I.34)
(I.34)
Отношение работы А, совершенной тепловой машиной, к количеству теплоты Q1, полученному от нагревателя, называется термодинамическим коэффициентом полезного действия (КПД) машины η:
![]() (I.35)
(I.35)
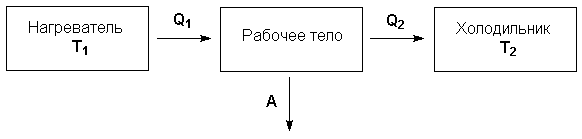
Рисунок 1.1 Схема
тепловой машины
Для получения математического выражения второго начала термодинамики рассмотрим работу идеальной тепловой машины (машины, обратимо работающей без трения и потерь тепла; рабочее тело – идеальный газ). Работа машины основана на принципе обратимого циклического процесса – термодинамического цикла Карно (рис. 1.2).
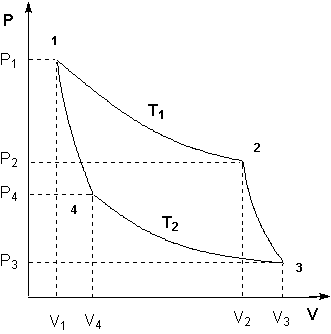
Рисунок 1.2 Цикл Карно
Запишем выражения для работы на всех участках цикла:
Участок 1 – 2: Изотермическое расширение.
![]() (I.36)
(I.36)
Участок 2 – 3: Адиабатическое расширение.
![]() (I.37)
(I.37)
Участок 3 – 4: Изотермическое сжатие.
![]() (I.38)
(I.38)
Участок 4 – 1: Адиабатическое сжатие.
![]() (I.39)
(I.39)
Общая работа в цикле равна сумме работ на всех участках:
![]() (I.40)
(I.40)
Проведя ряд несложных преобразований, получим для КПД идеальной тепловой машины, работающей по циклу Карно:
![]() (I.41)
(I.41)
Т.о., максимальный КПД тепловой машины не зависит от природы рабочего тела, а определяется только разностью температур нагревателя и холодильника. Очевидно, что без перепада температур превращение теплоты в работу невозможно. Полученное выражение справедливо для тепловой машины, обратимо работающей по любому циклу, поскольку любой цикл можно разбить на множество бесконечно малых циклов Карно.
Для необратимо работающей тепловой машины уравнение (I.41) преобразуется в неравенство:
![]() (I.42)
(I.42)
Для общего случая можем записать:
![]() (I.43)
(I.43)
На основе анализа работы идеальной тепловой машины Карно можно сделать следующий вывод, являющийся также одной из формулировок второго начала термодинамики:
Любая форма энергии может полностью перейти в теплоту, но теплота преобразуется в другие формы энергии лишь частично.
Т.о., можно условно принять, что внутренняя энергии системы состоит из двух составляющих: "свободной" X и "связанной" Y энергий, причем "свободная" энергия может быть переведена в работу, а "связанная" энергия может перейти только в теплоту.
![]() (I.44)
(I.44)
Величина связанной энергии тем больше, чем меньше разность температур, и при T = const тепловая машина не может производить работу. Мерой связанной энергии является новая термодинамическая функция состояния, называемая энтропией.
Введем определение энтропии, основываясь на цикле Карно. Преобразуем выражение (I.41) к следующему виду:
![]() (I.45)
(I.45)
Отсюда получаем, что для обратимого цикла Карно отношение количества теплоты к температуре, при которой теплота передана системе (т.н. приведенная теплота) есть величина постоянная:
![]() (I.46)
(I.46) ![]() (I.47)
(I.47)
Это верно для любого обратимого циклического процесса, т.к. его можно представить в виде суммы элементарных циклов Карно, для каждого из которых
![]() (I.48)
(I.48)
Т.о., алгебраическая сумма приведённых теплот для произвольного обратимого цикла равна нулю:
![]() (I.49)
(I.49)
Выражение (I.49) для любого цикла может быть заменено интегралом по замкнутому контуру:
![]() (I.50)
(I.50)
Если интеграл по замкнутому контуру равен нулю, то подынтегральное выражение есть полный дифференциал некоторой функции состояния; эта функция состояния есть энтропия S:
![]() (I.51)
(I.51)
Выражение (I.51) является определением новой функции состояния – энтропии и математической записью второго начала термодинамики для обратимых процессов. Если система обратимо переходит из состояния 1 в состояние 2, изменение энтропии будет равно:
![]() (I.52)
(I.52)
Подставляя (I.51, I.52) в выражения для первого начала термодинамики (I.1, I.2) получим совместное аналитическое выражение двух начал термодинамики для обратимых процессов:
![]() (I.53)
(I.53)
![]() (I.54)
(I.54)
Для необратимых процессов можно записать неравенства:
![]() (I.55)
(I.55)
![]() (I.56)
(I.56)
![]() (I.57)
(I.57)
Т.о., как следует из (I.57), работа обратимого процесса всегда больше, чем того же процесса, проводимого необратимо. Если рассматривать изолированную систему (δQ = 0), то легко показать, что для обратимого процесса dS = 0, а для самопроизвольного необратимого процесса dS > 0.
В изолированных системах самопроизвольно могут протекать только процессы, сопровождающиеся увеличением энтропии.
Энтропия изолированной системы не может самопроизвольно убывать.
Оба этих вывода также являются формулировками второго начала термодинамики.
1.4.1 Статистическая интерпретация энтропии
Классическая термодинамика рассматривает происходящие процессы безотносительно к внутреннему строению системы; поэтому в рамках классической термодинамики показать физический смысл энтропии невозможно. Для решения этой проблемы Больцманом в теорию теплоты были введены статистические представления. Каждому состоянию системы приписывается термодинамическая вероятность (определяемая как число микросостояний, составляющих данное макросостояние системы), тем большая, чем более неупорядоченным или неопределенным является это состояние. Т.о., энтропия есть функция состояния, описывающая степень неупорядоченности системы. Количественная связь между энтропией S и термодинамической вероятностью W выражается формулой Больцмана:
![]() (I.58)
(I.58)
С точки зрения статистической термодинамики второе начало термодинамики можно сформулировать следующим образом:
Система стремится самопроизвольно перейти в состояние с максимальной термодинамической вероятностью.
Статистическое толкование второго начала термодинамики придает энтропии конкретный физический смысл меры термодинамической вероятности состояния системы.
1.5 ТРЕТЬЕ НАЧАЛО ТЕРМОДИНАМИКИ
Ранее мы показали, что внутреннюю энергию системы можно условно представить в виде суммы двух величин "свободной" и "связанной" энергии. Возможность рассчитать величину "свободной" энергии, т.е. той части внутренней энергии системы, которую можно превратить в работу, дает тепловая теорема Нернста, называемая также третьим начало термодинамики.
Основные положения тепловой теоремы заключаются в следующем:
1. При абсолютном нуле температуры свободная энергия X равна теплоте процесса.
![]() (I.59)
(I.59)
2. При температурах, близких к абсолютному нулю, теплоемкость системы равна нулю.
![]() (I.60)
(I.60)
Одной из формулировок третьего начала термодинамики является также постулат Планка:
Энтропия идеального кристалла при абсолютном нуле температуры равна нулю.
Строго говоря, тепловая теорема Нернста и постулат
Планка являются следствиями из второго начала термодинамики; но независимо от
этого они имеют очень большое значение, позволяя рассчитывать абсолютную
энтропию системы и, следовательно, величину свободной энергии системы.
1.5.1 Расчет абсолютной энтропии
Рассчитаем изменение энтропии некоторой системы при нагревании её от абсолютного нуля до температуры T при постоянном давлении. Из первого и второго начал термодинамики имеем:
![]() (I.61)
(I.61)
![]() (I.62)
(I.62)
Отсюда:
![]() (I.63)
(I.63)
Учитывая, что ST=0 = 0, получим:
![]() (I.64)
(I.64)
При T = 0 любое вещество может находиться только в твердом состоянии. При нагревании вещества возможен его переход в жидкое и затем в газообразное состояние; для фазовых переходов, происходящих в изобарно-изотермических условиях, изменение энтропии равно приведенной теплоте фазового перехода:
![]() (I.65)
(I.65)
Таким образом, нагревание вещества без фазовых переходов сопровождается непрерывным ростом энтропии; при фазовом переходе происходит скачкообразное изменение энтропии. Графическая зависимость энтропии вещества от температуры приведена на рисунке 1.3.
Учитывая это, рассчитать абсолютную энтропию любого вещества при любой температуре можно следующим образом:
![]() (I.66)
(I.66)
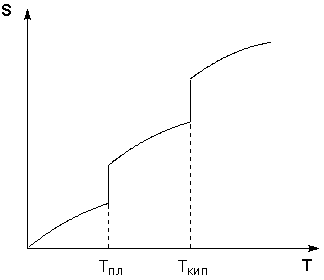
Рис. 1.3 Зависимость энтропии вещества от температуры.
Поскольку энтропия есть функция состояния, изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции; оно может быть рассчитано по уравнению (I.67):
![]() (I.67)
(I.67)
Для многих веществ величины абсолютной энтропии в стандартных условиях приведены в справочной литературе.
1.6 ТЕРМОДИНАМИЧЕСКИЕ ПОТЕНЦИАЛЫ
Изменение энтропии однозначно определяет направление и предел самопроизвольного протекания процесса лишь для наиболее простых систем – изолированных. На практике же большей частью приходится иметь дело с системами, взаимодействующими с окружающей средой. Для характеристики процессов, протекающих в закрытых системах, были введены новые термодинамические функции состояния: изобарно-изотермический потенциал (свободная энергия Гиббса) и изохорно-изотермический потенциал (свободная энергия Гельмгольца).
Поведение всякой термодинамической системы в общем случае определяется одновременным действием двух факторов – энтальпийного, отражающего стремление системы к минимуму тепловой энергии, и энтропийного, отражающего противоположную тенденцию – стремление системы к максимальной неупорядоченности. Если для изолированных систем (ΔН = 0) направление и предел самопроизвольного протекания процесса однозначно определяется величиной изменения энтропии системы ΔS, а для систем, находящихся при температурах, близких к абсолютному нулю (S = 0 либо S = const) критерием направленности самопроизвольного процесса является изменение энтальпии ΔН, то для закрытых систем при температурах, не равных нулю, необходимо одновременно учитывать оба фактора. Направлением и предел самопроизвольного протекания процесса в любых системах определяет более общий принцип минимума свободной энергии:
Самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения.
Для закрытых систем, находящихся в изобарно-изотермических либо изохорно-изотермических условиях свободная энергия принимает вид изобарно-изотермического либо изохорно-изотермического потенциалов (т.н. свободная энергия Гиббса и Гельмгольца соответственно). Данные функции называют иногда просто термодинамическими потенциалами, что не вполне строго, поскольку термодинамическими потенциалами являются также внутренняя энергия (изохорно-изэнтропный) и энтальпия (изобарно-изэнтропный потенциал).
Рассмотрим закрытую систему, в которой осуществляется равновесный процесс при постоянных температуре и объеме. Выразим работу данного процесса, которую обозначим Amax (поскольку работа процесса, проводимого равновесно, максимальна), из уравнений (I.53, I.54):
![]() (I.68)
(I.68)
![]() (I.69)
(I.69)
Преобразуем выражение (I.69), сгруппировав члены с одинаковыми индексами:
![]() (I.70)
(I.70)
Введя обозначение:
![]() (I.71)
(I.71)
получаем:
![]() (I.72)
(I.72)
![]() (I.73)
(I.73)
Функция ![]() есть изохорно-изотермический
потенциал (свободная энергия Гельмгольца), определяющий направление и
предел самопроизвольного протекания процесса в закрытой системе, находящейся в
изохорно-изотермических условиях.
есть изохорно-изотермический
потенциал (свободная энергия Гельмгольца), определяющий направление и
предел самопроизвольного протекания процесса в закрытой системе, находящейся в
изохорно-изотермических условиях.
Закрытую систему, находящуюся в изобарно-изотермических условиях, характеризует изобарно-изотермический потенциал G:
![]() (I.74)
(I.74)
![]() (I.75)
(I.75)
Поскольку –ΔF = Amax, можно записать:
![]() (I.76)
(I.76)
Величину А'max называют максимальной
полезной работой (максимальная работа за вычетом работы расширения).
Основываясь на принципе минимума свободной энергии, можно сформулировать
условия самопроизвольного протекания процесса в закрытых системах.
Условия самопроизвольного протекания процессов в закрытых системах:
Изобарно-изотермические (P = const, T = const):
ΔG < 0, dG < 0
ΔF < 0, dF < 0
Процессы, которые сопровождаются увеличением термодинамических потенциалов, протекают лишь при совершении работы извне над системой. В химии наиболее часто используется изобарно-изотермический потенциал, поскольку большинство химических (и биологических) процессов происходят при постоянном давлении. Для химических процессов величину ΔG можно рассчитать, зная ΔH и ΔS процесса, по уравнению (I.75), либо пользуясь таблицами стандартных термодинамических потенциалов образования веществ ΔG°обр; в этом случае ΔG° реакции рассчитывается аналогично ΔН° по уравнению (I.77):
![]() (I.77)
(I.77)
Величина стандартного изменения изобарно-изотермического потенциала в ходе химической любой реакции ΔG°298 есть мера химического сродства исходных веществ. Основываясь на уравнении (I.75), можно оценить вклад энтальпийного и энтропийного факторов в величину ΔG и сделать некоторые обобщающие заключения о возможности самопроизвольного протекания химических процессов, основываясь на знаке величин ΔН и ΔS.
1. Экзотермические реакции; ΔH < 0.
а) Если ΔS > 0, то ΔG всегда отрицательно; экзотермические реакции, сопровождающиеся увеличением энтропии, всегда протекают самопроизвольно.
б) Если ΔS < 0, реакция будет идти самопроизвольно при ΔН > TΔS (низкие температуры).
2. Эндотермические реакции; ΔH > 0.
а) Если ΔS > 0, процесс будет самопроизвольным при ΔН < TΔS (высокие температуры).
б) Если ΔS < 0, то ΔG всегда положительно; самопроизвольное протекание эндотермических реакций, сопровождающихся уменьшением энтропии, невозможно.
1.7 ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Как было показано выше, протекание самопроизвольного процесса в термодинамической системе сопровождается уменьшением свободной энергии системы (dG < 0, dF < 0). Очевидно, что рано или поздно (напомним, что понятие "время" в термодинамике отсутствует) система достигнет минимума свободной энергии. Условием минимума некоторой функции Y = f(x) является равенство нулю первой производной и положительный знак второй производной: dY = 0; d2Y > 0. Таким образом, условием термодинамического равновесия в закрытой системе является минимальное значение соответствующего термодинамического потенциала:
Изобарно-изотермические (P = const, T = const):
ΔG = 0 dG = 0, d2G > 0
Изохорно-изотермические (V = const, T = const):
ΔF = 0 dF = 0, d2F > 0
Состояние системы с минимальной свободной энергией есть состояние термодинамического равновесия:
Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом.
Учение о равновесных состояниях – один из разделов термодинамики. Далее мы будем рассматривать частный случай термодинамического равновесного состояния – химическое равновесие. Как известно, многие химические реакции являются обратимыми, т.е. могут одновременно протекать в обоих направлениях – прямом и обратном. Если проводить обратимую реакцию в закрытой системе, то через некоторое время система придет в состояние химического равновесия – концентрации всех реагирующих веществ перестанут изменяться во времени. Необходимо отметить, что достижение системой состояния равновесия не означает прекращения процесса; химическое равновесие является динамическим, т.е. соответствует одновременному протеканию процесса в противоположных направлениях с одинаковой скоростью. Химическое равновесие является подвижным – всякое бесконечно малое внешнее воздействие на равновесную систему вызывает бесконечно малое изменение состояния системы; по прекращении внешнего воздействия система возвращается в исходное состояние. Ещё одним важным свойством химического равновесия является то, что система может самопроизвольно прийти в состояние равновесия с двух противоположных сторон. Иначе говоря, любое состояние, смежное с равновесным, является менее устойчивым, и переход в него из состояния равновесия всегда связан с необходимостью затраты работы извне.
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции
![]()
соответствующие константы равновесия выражаются следующим образом:
![]() (I.78)
(I.78) ![]() (I.79)
(I.79)
![]() (I.80)
(I.80)
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений.
Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
![]() (I.81)
(I.81)
![]() (I.82)
(I.82)
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие.
Приняв, что V1 = V2, можно записать:
![]() (I.83)
(I.83)
![]() (I.84)
(I.84)
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л.
Теперь рассмотрим (с некоторыми упрощениями) более строгий термодинамический вывод выражения для константы равновесия. Для этого необходимо ввести понятие химический потенциал. Очевидно, что величина свободной энергии системы будет зависеть как от внешних условий (T, P или V), так и от природы и количества веществ, составляющих систему. В случае, если состав системы изменяется во времени (т.е. в системе протекает химическая реакция), необходимо учесть влияние изменения состава на величину свободной энергии системы. Введем в некоторую систему бесконечно малое количество dni молей i-го компонента; это вызовет бесконечно малое изменение термодинамического потенциала системы. Отношение бесконечно малого изменения величины свободной энергии системы к бесконечно малому количеству компонента, внесенному в систему, есть химический потенциал μi данного компонента в системе:
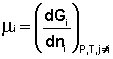 (I.85)
(I.85)
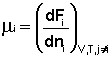 (I.86)
(I.86)
Химический потенциал компонента связан с его парциальным давлением или концентрацией следующими соотношениями:
![]() (I.87)
(I.87)
![]() (I.88)
(I.88)
Здесь μ°i – стандартный химический потенциал компонента (Pi = 1 атм., Сi = 1 моль/л.). Очевидно, что изменение свободной энергии системы можно связать с изменением состава системы следующим образом:
![]() (I.89)
(I.89)
![]() (I.90)
(I.90)
Поскольку условием равновесия является минимум свободной энергии системы (dG = 0, dF = 0), можно записать:
![]() (I.91)
(I.91)
В закрытой системе изменение числа молей одного компонента сопровождается эквивалентным изменением числа молей остальных компонентов; т.е., для приведенной выше химической реакции имеет место соотношение:
![]() (I.92)
(I.92)
Отсюда можно получить следующее условие химического равновесия в закрытой системе:
![]() (I.93)
(I.93)
В общем виде условие химического равновесия можно записать следующим образом:
![]() (I.94)
(I.94)
Выражение (I.94) носит название уравнения Гиббса – Дюгема. Подставив в него зависимость химического потенциала от концентрации, получаем:
![]() (I.95)
(I.95)
Поскольку Σniμi = ΔF, а Σniμ°i = ΔF°, получаем:
![]() (I.96)
(I.96)
Для изобарно-изотермического процесса аналогичным образом можно получить:
![]() (I.97)
(I.97)
Полученные нами выражения I.96 – I.97 есть изотерма химической реакции. Если система находится в состоянии химического равновесия, то изменение термодинамического потенциала равно нулю; получаем:
![]() (I.98)
(I.98)
![]() (I.99)
(I.99)
Здесь сi и рi – равновесные концентрации и парциальные давления исходных веществ и продуктов реакции (в отличие от неравновесных Сi и Рi в уравнениях I.96 – I.97).
Поскольку для каждой химической реакции стандартное изменение термодинамического потенциала ΔF° и ΔG° есть строго определенная величина, то произведение равновесных парциальных давлений (концентраций), возведенных в степень, равную стехиометрическому коэффициенту при данном веществе в уравнении химической реакции (стехиометрические коэффициенты при исходных веществах принято считать отрицательными) есть некоторая константа, называемая константой равновесия. Уравнения (I.98, I.99) показывают связь константы равновесия со стандартным изменением свободной энергии в ходе реакции. Уравнение изотермы химической реакции связывает величины реальных концентраций (давлений) реагентов в системе, стандартного изменения термодинамического потенциала в ходе реакции и изменения термодинамического потенциала при переходе из данного состояния системы в равновесное. Знак ΔG (ΔF) определяет возможность самопроизвольного протекания процесса в системе. При этом ΔG° (ΔF°) равно изменению свободной энергии системы при переходе из стандартного состояния (Pi = 1 атм., Сi = 1 моль/л) в равновесное. Уравнение изотермы химической реакции позволяет рассчитать величину ΔG (ΔF) при переходе из любого состояния системы в равновесное, т.е. ответить на вопрос, будет ли химическая реакция протекать самопроизвольно при данных концентрациях Сi (давлениях Рi) реагентов:
![]() (I.100)
(I.100)
![]() (I.101)
(I.101)
Если изменение термодинамического потенциала меньше
нуля, процесс в данных условиях будет протекать самопроизвольно.
1.7.1 Влияние внешних условий на химическое равновесие
При постоянстве внешних условий система может находиться в состоянии равновесия сколь угодно долго. Если изменить эти условия (т.е. оказать на систему какое-либо внешнее воздействие), равновесие нарушается; в системе возникает самопроизвольный процесс, который продолжается до тех пор, пока система опять не достигнет состояния равновесия (уже при новых условиях). Рассмотрим, как влияют на положение равновесия некоторые факторы.
1.7.2 Влияние давления и концентрации
Рассмотрим несколько возможных случаев смещения равновесия.
1. В систему добавлено исходное вещество. В этом случае
![]() ;
; ![]() ;
;
По уравнению изотермы химической реакции (I.100 – I.101) получаем: ΔF < 0; ΔG < 0. В системе возникнет самопроизвольный химический процесс, направленный в сторону расходования исходных веществ и образования продуктов реакции (химическое равновесие смещается вправо).
2. В систему курсовые - 700 р.
Работы, похожие на Учебное пособие: Коллоидная химия
Общая и неорганическая химия
Квантово-механическая модель атома. Квантовые числа. Атомные орбитали. Порядок заполнения орбиталей электронами Теория строения атома основана на ...
Процесс диссоциации слабых электролитов является обратимым и в системе существует динамическое равновесие, которое может быть описано константой равновесия, выраженной через ...
В результате того, что в растворе образуется сильный электролит гидроксид калия, концентрация гидроксид-ионов ОН- будет значительно больше концентрации ионов водорода Н+. В ...
Раздел: Рефераты по химии
Тип: учебное пособие
Методы анализа лекарственных препаратов
Оглавление Вступление Глава 1. Основные принципы фармацевтического анализа 1.1 Критерии фармацевтического анализа 1.2 Ошибки, возможные при проведении ...
Для выполнения испытаний используют буферные растворы с постоянной концентрацией водородных ионов, отличающихся друг от друга на величину рН, равную 0,2. К серии таких растворов и ...
В процессе титрования в растворе уменьшается концентрация йодида калия, равновесие смещается влево, уменьшается концентрация йода, и синяя окраска исчезает Возможность определения ...
Раздел: Рефераты по медицине
Тип: дипломная работа
Химический состав молока
ВВЕДЕНИЕ Химия и физика как наука начала свой отсчет в прошлом веке, в тот период она начинала с изучения химического состава молока. В нашей стране ...
Молоко содержит ряд химических соединений, способных отдавать или присоединять электроны (атомы Н2): аскорбиновую кислоту (токоферолы), цистеин, рибофлавин, молочную кислоту ...
Молочная сыворотка - это реальный раствор, в противоположность идеальному раствору, которые практически реализуется только при бесконечном разбавлении и в котором растворенные ...
Раздел: Биология и химия
Тип: курсовая работа
Получение гидроксида натрия каустификацией содового раствора
Введение Гидроксид натрия (каустическая сода) используется во многих отраслях промышленности: химической, металлургической, нефтеперерабатывающей ...
Процесс каустификации содового раствора служит характерным примером обратимого взаимодействия, идущего в кинетической области, в системе жидкость - твердое (Ж-Т). Ход реакции (5 ...
Скорость межфазных процессов обычно велика в начале контакта фаз и постепенно уменьшается во времени до некоторого постоянного значения, характеризуемого константой химического ...
Раздел: Рефераты по химии
Тип: учебное пособие
Коррозия меди в 5М изопропанольных растворах НС1
МИНИСТЕРСТВО ОБЩЕГО И ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ Тамбовский государственный университет им. Г.Р. Державина КАФЕДРА ...
При малых концентрациях FeC13 главную роль играет толщина поверхностного слоя, а при высоких концентрациях FeC13 - диффузия ионов Fe3+ в твердую фазу.
Уменьшение экранирования поверхности электрода происходит при интенсивном перемешивании, снижение концентрации Fe3+ - ионов и повышение концентрации С1- -ионов, которые, по ...
Раздел: Рефераты по химии
Тип: реферат