Дипломная работа: Сравнительный анализ: методы получения синтез-газа
Министерство образования Российской Федерации
Московская государственная академия тонкой химической
технологии им. М.В. Ломоносова
Кафедра Технологии нефтехимического синтеза
и искусственного жидкого топлива
АТТЕСТАЦИОННАЯ РАБОТА
на соискание степени бакалавра по направлению
550800 «Химическая технология и биотехнология»
Тема: Сравнительный анализ: методы получения синтез-газа
Заведующий кафедрой,
д. х. н., проф. Третьяков В. Ф.
Руководитель,
ст. преп. Антонюк С. Н.
Дипломант, студент группы ХТ-406
Сысоев М. М.
Москва, 2003 г.
Содержание
1. Введение
2. Способы получения синтез-газа
3. Газификация угля
3.1 Тенденции развития и новые инженерные решения в газификации угля
3.2 Взгляд на углепереработку сквозь десятилетия
3.3 Инженерные разработки за прошедшее столетие
3.4 Аппаратурно-техническое оформление процесса
4. Конверсия метана в синтез-газ
4.1 Термодинамика процесса
4.2 Кинетика углекислотной конверсии метана
4.3 Механизм конверсии смеси CH4 + CO2
4.4 Катализаторы углекислотной конверсии метана
4.5 Технология конверсии метана
5. Синтез Фишера-Тропша
5.1 Выбор катализаторов
6. Альтернативный способ получения синтез-газа
6.1 Термохимическая конверсия биомассы
6.2 Биотехнологическая конверсия биомассы
7. Продукты, получаемые на основе синтез-газа
8. Выводы
9. Используемая литература
1. Введение
История знает немало примеров, когда в силу острой необходимости рождались новые оригинальные подходы к решению давно существующих жизненно важных проблем. Так, в предвоенной Германии, лишенной доступа к нефтяным источникам, назревал жесткий дефицит топлива, необходимого для функционирования мощной военной техники. Располагая значительными запасами ископаемого угля, Германия была вынуждена искать пути его превращения в жидкое топливо. Эта проблема была успешно решена усилиями превосходных химиков, из которых, прежде всего следует упомянуть Франца Фишера, директора Института кайзера Вильгельма по изучению угля.
В 1926 году была опубликована работа Ф. Фишера и Г. Тропша "О прямом синтезе нефтяных углеводородов при обыкновенном давлении", в которой сообщалось, что при восстановлении водородом монооксида углерода при атмосферном давлении в присутствии различных катализаторов (железо - оксид цинка или кобальт - оксид хрома) при 270 оС получаются жидкие и даже твердые гомологи метана.
Так возник знаменитый синтез углеводородов из монооксида углерода и водорода, называемый с тех пор синтезом Фишера-Тропша. Смесь CO и H2 в различных соотношениях, называемая синтез-газом, легко может быть получена как из угля, так и из любого другого углеродсодержащего сырья.
Следует отметить, что к моменту разработки синтеза Фишера-Тропша существовал другой способ получения жидкого топлива - не из синтез-газа, а непосредственно из угля прямой гидрогенизацией. В этой области значительных успехов добился также немецкий химик Ф. Бергиус, который в 1911 году получил из угля бензин. Справедливости ради подчеркнем, что синтез Фишера-Тропша возник не на пустом месте - к тому времени существовали научные предпосылки, которые базировались на достижениях органической химии и гетерогенного катализа. Еще в 1902 году П. Сабатье и Ж. Сандеран впервые получили метан из СО и H2 . В 1908 году Е. Орлов открыл, что при пропускании монооксида углерода и водорода над катализатором, состоящим из никеля и палладия, нанесенных на уголь, образуется этилен.
Промышленность искусственного жидкого топлива достигла наибольшего подъема в годы второй мировой войны. Достаточно сказать, что синтетическое топливо почти полностью покрывало потребности Германии в авиационном бензине. После 1945 года в связи с бурным развитием нефтедобычи и падением цен на нефть отпала необходимость синтеза жидких топлив из СО и Н2 . Наступил нефтехимический бум. Однако в 1973 году разразился нефтяной кризис - нефтедобывающие страны ОПЕК (Организация стран - экспортеров нефти - Organization of Petroleum Exporting Countries) резко повысили цены на сырую нефть, и мировое сообщество вынуждено было осознать реальную угрозу истощения в обозримые сроки дешевых и доступных нефтяных ресурсов. Энергетический шок 70-х годов возродил интерес ученых и промышленников к использованию альтернативного нефти сырья, и здесь первое место, бесспорно, принадлежит углю. Мировые запасы угля огромны, они, по различным оценкам, более чем в 50 раз превосходят нефтяные ресурсы, и их может хватить на сотни лет. Нет никаких сомнений, что в обозримом будущем использование синтез-газа будет играть ключевую роль не только и не столько для производства "угольных" топлив (здесь трудно пока конкурировать с нефтяным топливом), но, прежде всего для целей органического синтеза. В настоящее время в промышленном масштабе по методу Фишера-Тропша получают бензин, газойль и парафины только в Южной Африке. На установках фирмы "Sasol" производят около 5 млн. т. в год жидких углеводородов.
Отражением интенсификации исследований по синтезам на основе СО и Н2 является резкое возрастание публикаций, посвященных химии одноуглеродных молекул (так называемая С1-химия). С 1984 года начал издаваться международный журнал "C1-Molecule Chemistry". Таким образом, мы являемся свидетелями наступающего ренессанса в истории углехимии. Рассмотрим некоторые пути превращения синтез-газа, приводящие к получению как углеводородов, так и некоторых ценных кислородсодержащих соединений. Важнейшая роль в превращениях СО принадлежит гетерогенному и гомогенному катализу [1-3].
2. Способы получения синтез-газа
Первым способом получения синтез-газа была газификация каменного угля, которая была осуществлена еще в 30-е годы XIX века в Англии с целью получения горючих газов: водорода, метана, монооксида углерода. Этот процесс широко использовался во многих странах до середины 50-х годов XX века, а затем был вытеснен методами, основанными на использовании природного газа и нефти. Однако в связи с сокращением нефтяных ресурсов значение процесса газификации снова стало возрастать.
В настоящее время существуют три основных промышленных метода получения синтез-газа[34].
1. Газификация угля. Процесс основан на взаимодействии угля с водяным паром:
C + H2O ↔ H2 + CO
Эта реакция является эндотермической, равновесие сдвигается вправо при температурах 900-1000 оС. Разработаны технологические процессы, использующие парокислородное дутье, при котором наряду с упомянутой реакцией протекает экзотермическая реакция сгорания угля, обеспечивающая нужный тепловой баланс:
C + 1/2O2↔CO
2. Конверсия метана. Реакция взаимодействия метана с водяным паром проводится в присутствии никелевых катализаторов (Ni-Al2O3) при повышенных температурах (800-900 оС) и давлении:
CH4 + H2O → CO + 3H2
В качестве сырья вместо метана может быть использовано любое углеводородное сырье.
3. Парциальное окисление углеводородов. Процесс заключается в неполном термическом окислении углеводородов при температурах выше 1300 оС:
CnH2n + 2 + 1/2nO2 → nCO + (n + 1)H2
Способ применим к любому углеводородному сырью, но наиболее часто в промышленности используют высококипящую фракцию нефти - мазут.
Соотношение СО : Н2 существенно зависит от применяемого способа получения синтез-газа. При газификации угля и парциальном окислении это соотношение близко к 1 : 1, тогда как при конверсии метана соотношение СО : Н2 составляет 1 : 3. В настоящее время разрабатываются проекты подземной газификации, то есть газификации угля непосредственно в пласте. Интересно, что эта идея была высказана Д.И. Менделеевым более 100 лет назад. В перспективе синтез-газ будут получать газификацией не только угля, но и других источников углерода вплоть до городских и сельскохозяйственных отходов.
3. Газификация угля
Газификация — высокотемпературный процесс взаимодействия углерода топлива с окислителями, проводимый с целью получения горючих газов (Н2, СО, СН4). В качестве окислителей, которые иногда называют газифицирующими агентами, используют кислород (или обогащенный им воздух), водяной пар, диоксид углерода либо смеси указанных веществ. В зависимости от соотношения исходных реагентов, температуры, продолжительности реакции и других факторов можно получать газовые смеси самого разного состава.
3.1 Тенденции развития и новые инженерные решения в газификации угля
Впервые промышленная реализация газификации твердых топлив была осуществлена в 1835 г в Великобритании. К 50-м годам XIX в. практически во всех крупных и средних городах Европы и Северной Америки действовали газовые заводы для производства отопительного, бытового и светильного газа [4]. К середине XX в этот процесс получил широкое развитие в большинстве промышленных стран мира. Например, в СССР в 50-е годы работало свыше 350 газогенераторных станций, на которых было установлено около 2500 газогенераторов. Эти станции вырабатывали ежегодно 35 млрд. м3 энергетических и технологических газов. Это был "золотой век" газификации угля. Начиная с 60-х годов XIX в., все более серьезную конкуренцию углю начинает оказывать нефть. В начале 1960-х годов разработка месторождений дешевой нефти на Ближнем Востоке и в Западной Сибири привела практически к полной ликвидации этой отрасли промышленности. Как известно, в последующие 20—25 лет в мировом энергетическом балансе происходили изменения, обусловленные ростом добычи и потребления нефти, попутных и природных газов. Вследствие этого конкурентоспособность искусственных энергетических и технологических газов, получаемых из твердых топлив, резко снизилась, и их производство практически повсеместно было прекращено. Сохранились лишь небольшие островки в уникальных регионах. Например, в ЮАР углепереработка (главным образом на основе газификации угля) стала крупной промышленным сектором из-за эмбарго на поставку нефти. Началось триумфальное шествие нефти. Однако уже в 1972 г. оно омрачилось первым "энергетическим кризисом", который по существу был спровоцирован на политической основе странами-участниками ОПЕК. Мировые цены на нефть подскочили с 5-7 до 24 долл. США за баррель (1 т сырой нефти сорта Brent ≈ 8,06 баррелей), и стало ясно, что углепереработку списывать в архив рано, так как в большинстве развитых стран много угля и мало или совсем нет нефти.
Этот кризис преподнес цивилизованному миру очень важный урок. Во-первых, все осознали, что запасы углеводородного сырья распределены крайне неравномерно и неудобно, и, во-вторых, эти запасы - исчерпаемы. Запасы же угля и других твердых горючих ископаемых – нефтяных сланцев, битумных песков, торфа и т.п. распределены более равномерно, и сроки их исчерпания оценивается многими сотнями лет. Но самый главный результат этот кризиса заключается в активизации работ по энергосбережению.
Однако в последние годы в связи с сокращением ресурсов нефтяного и газового сырья процесс газификации твердых горючих ископаемых вновь привлек к себе внимание, искусственные газы опять начинают рассматриваться как одна из существенных составляющих теплового баланса. Например, в США планировалось к 1990 г построить 63 завода этого профиля средней мощностью ~7 млн. м3 газа в сутки каждый. Их годовая выработка составляла 140 млрд. м3, а к 2000 г увеличилась до 220—250 млрд. м3, что соответствует ~23% потребности США в энергетических и технологических газах.
3.2 Взгляд на углепереработку сквозь десятилетия
В середине 1980-х годов интерес к углепереработке пошел на убыль. Причин несколько.
Во-первых, политикой "кнута и пряника" США установили контроль над странами - производителями нефти. Наиболее амбициозных (Ирак, Иран) наказали в назидание другим. В результате рост цен на нефть замедлился. Сохранять равновесие поручили шестому флоту США и силам быстрого реагирования. Насколько это равновесие устойчиво покажет время.
В течение 1980-х годов цены на нефть снизились с 40 долл. США за баррель (что соответствует примерно 65 долл. США за баррель в современных ценах с поправкой на инфляцию) до минимального уровня 9,13 долл. США за баррель в декабре 1998 г. и в настоящее время колеблются в "коридоре" 17-27 долл. США за баррель.
Во-вторых, эффективно сработали государственные программы энергосбережения, что в конечном итоге привело к снижению темпа роста потребления нефти и природного газа. С середины 1970-х годов энергоемкость единицы ВВП в развитых странах снизилась на 22 %, а нефтеемкость на 38 % [5].
В-третьих, динамичное развитие нефтегазовой отрасли и масштабные работы по разведке новых месторождений нефти и газа показали, что запасы углеводородного сырья на самом деле значительно больше, чем предполагалось. Последние 20 лет ежегодный прирост разведанных запасов нефти и газа опережает их потребление, и прогнозные сроки исчерпания регулярно отодвигаются. По достаточно авторитетным данным глобальную замену нефти углем следует ожидать после середины XXI в., а замену природного газа углем – к концу века. Если, конечно, не произойдет прорыва в развитии технологии ядерного синтеза.
В-четвертых, ни одна из разрабатываемых технологий не позволила повысить рентабельность процесса получения жидкого топлива из угля в такой степени, чтобы "синтетическая нефть" могла конкурировать с природной нефтью.
В итоге “эпоха угля” не наступила и интерес к переработке угля уменьшился. Большинство программ было свернуто, а оставшиеся - радикально урезаны. Более десятка проектов были завершены на стадии 5-летней готовности, т.е. при изменении конъюнктуры рынка углеводородного сырья можно в течение 5 лет на основе демонстрационных установок производительностью 10-60 т/ч по углю развернуть промышленное производство. Если от коммерческого использования технологий прямого и непрямого ожижения угля в конце 1980-х гг. пока отказались, то интерес к газификации угля хотя и уменьшился, но не прекратился. Например, в ряде регионов, где природного газа нет или мало (Северная Америка, Китай и др.), использование газа из угля для синтеза метанола и аммиака экономически оправдано и построен ряд промышленных предприятий.
В 1990-е годы бурное развитие получила внутрицикловая газификация для производства электроэнергии, т.е. использование бинарного цикла, при котором горючий газ утилизируется в газовой турбине, а продукты сгорания используются при генерации пара для паровой турбины. Первая коммерческая электростанция с внутрицикловой газификацией – Cool Water, США, шт. Калифорния, мощностью 100 МВт (60 т/ч по углю) была построена в 1983 г. Использовался газогенератор Texaco с подачей топлива в виде водо-угольной суспензии. После 1993 г. в разных странах было введено в эксплуатацию 18 электростанций с внутри цикловой газификацией твердого топлива мощностью от 60 до 300 МВт. На рис.1 приведены данные по мировому производству газа из твердых топлив с 1970 г., а в табл. 1 – структура его потребления.
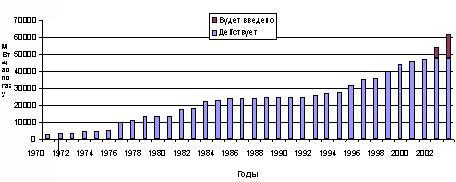
Рис. 1. Суммарная мощность газогенераторных установок
Таблица 1
Динамика потребления газа из угля в мире
| Целевое использование | Использование в 2001 г., МВт по газу | Доля в 2001 г., % | Вводится в эксплуатацию до конца 2004 г., МВт по газу | Годовой прирост мощности в 2002-2004 гг., % |
| Химическое производство | 18 000 | 45 | 5 000 | 9,3 |
| Внутрицикловая газификация (производство электроэнергии) | 12 000 | 30 | 11 200 | 31 |
| Синтез по Фишеру-Тропшу | 10 000 | 25 | 0 | 0 |
| ВСЕГО | 40 000 | 100 | 17 200 | 14,3 |
Приведенные данные наглядно демонстрируют ускорение динамики вовлечения газификации угля в мировую промышленность. Повышенный интерес к внутрицикловой газификации угля в развитых странах объясняется двумя причинами.
Во-первых, ТЭС с внутрицикловой газификацией экологически менее опасна. Благодаря предварительной очистке газа сокращаются выбросы оксидов серы, азота и твердых частиц.
Во-вторых, использование бинарного цикла позволяет существенно увеличить КПД электростанции и, следовательно, сократить удельный расход топлива.
В табл. 2 приведены характерные величины удельных выбросов и КПД для ТЭС с внутрицикловой газификацией и для ТЭС с традиционным сжиганием угля.
Таблица 2
Величины удельных выбросов и КПД для ТЭС с внутрицикловой газификацией и с традиционным сжиганием угля
| Параметры | Традиционная угольная ТЭС | ТЭС с внутрицикловой газификацией |
|
Концентрация вредных
веществ в дымовых газах |
130 |
10 |
| Электрический КПД, % | 33-35 | 42-46 |
Необходимо отметить, что удельные капитальные затраты при использовании внутрицикловой газификации составляют примерно 1500 долл. США за 1кВт с перспективой снижения до 1000-1200 долл. США, в то время как для традиционной угольной ТЭС удельные капитальные затраты составляют примерно 800-900 долл. США за 1 кВт. Ясно, что ТЭС с внутрицикловой газификацией твердого топлива более привлекательна при наличии экологических ограничений в месте размещения и при использовании достаточно дорогого топлива, так как расход топлива на 1 кВт сокращается.
Эти условия характерны для развитых стран. В настоящее время использование внутрицикловой газификации твердого топлива считается самым перспективным направлением в энергетике.
3.3 Инженерные разработки за прошедшее столетие
В настоящее время выявились следующие наиболее экономически эффективные области применения метода газификации:
- газификация сернистых и многозольных топлив с последующим сжиганием полученных газов на мощных тепловых электростанциях. В углях, ежегодно добываемых в России, содержится около 10 млн. т серы, большая часть которой при сжигании выбрасывается в атмосферу в виде токсичных оксидов серы и серооксида углерода. При газификации сернистых углей образуется сероводород, который можно сравнительно легко извлечь и затем переработать в товарную серу или серную кислоту
- газификация твердых топлив для крупномасштабного производства заменителей природного газа. Это направление имеет наибольшее значение для местного газоснабжения районов, удаленных от месторождений природного газа и нефти или от магистральных трубопроводов
- газификация твердых топлив с целью получения синтез-газа, газов-восстановителей и водорода для нужд химической, нефтехимической и металлургической промышленности.
Процесс газификации зависит от многих факторов, влияющих на состав получаемого газа и его теплоту сгорания. В связи с этим до сих пор отсутствует единая общепринятая классификация методов осуществления рассматриваемого процесса. Ниже приведен один из возможных вариантов классификации.
· по виду дутья (газифицирующего агента): воздушное, воздушно-кислородное, паровоздушное, парокислородное.
· по давлению: при атмосферном давлении, при повышенном давлении.
· по размеру частиц топлива: газификация крупнозернистого (кускового), мелкозернистого и пылевидного топлива.
· по конструктивным особенностям реакционной зоны: в неподвижном плотном слое топлива, в псевдоожиженном слое топлива, в пылеугольном факеле.
· по способу выведения золы: в твердом виде, в виде жидкого шлака.
· по способу подвода тепла: при частичном сжигании топлива в газогенераторе, при смешении топлива с предварительно нагретым твердым, жидким или газообразным теплоносителем (регенеративный нагрев), при подводе тепла через стенку аппарата (рекуперативный нагрев).
· по назначению получаемого газа: получение газов с заданной теплотой сгорания (низкой — до 6700 кДж/м3, средней — от 12000 до 18000 кДж/м3 и высокой — от 30000 до 35000 кДж/м3); получение газов заданного состава.
· по способу обогащения конечного газа метаном: безостаточная газификация топлива в СО, СО2 и Н2 в сочетании с отдельной стадией метанирования СО и СО2 водородом; газификация с полным выделением летучих и максимальным образованием метана в слое топлива; гидрогазификация.
Газификации может быть подвергнуто большинство известных видов твердых горючих ископаемых. При этом можно получить газ заданного состава или заданной теплоты сгорания, так как эти показатели в значительной степени определяются температурой, давлением и составом применяемого дутья.
Газ с низкой теплотой сгорания образуется при использовании воздушного или паровоздушного дутья. В соответствии с этим его называют воздушным или паровоздушным (смешанным). Он характеризуется высоким содержанием балласта — азота (до 40—50% об.), что обусловливает низкую теплоту сгорания такого газа. Основная область применения таких газов — сжигание в топках промышленных печей. Кроме того, после, конверсии содержащегося в них оксида углерода и очистки от СО2 получают азотоводородную смесь — исходное сырье для синтеза аммиака.
Газы со средней теплотой сгорания получают в процессах паровой или парокислородной газификации твердых топлив под давлением до 2—2,5 МПа. По составу они представляют собой смеси оксидов углерода и водорода с небольшими количествами метана и других углеводородов: 30—35% (об.) СО2, 10—13% (об.) СО, 38—40% (об.) Н2) 10—12% (об.) СН4, 0,5— 1,5% (об.) СnН2n. По экономическим соображениям такие газы применяют в ограниченных масштабах. Их используют главным образом как химическое сырье, а также начинают применять в металлургии в качестве газов-восстановителей.
Технология получения указанных газов первоначально была основана на использовании паровоздушного дутья, причем воздух предварительно обогащался кислородом до 40% (об.). Наряду с этим повысить теплоту сгорания газа можно, проводя газификацию при повышенном давлении. Другой способ получения газов со средней теплотой сгорания — газификация твердых топлив с применением парового дутья и предварительно нагретого до 900—1100 °С твердого теплоносителя. В качестве последнего можно использовать золу, остающуюся после сжигания части топлива в выносной топке. Подобный вариант позволяет получать газ, состоящий в основном из СО и Н2 в соотношении, близком к 1:1, однако этот способ опробован пока лишь на небольших опытно-промышленных установках.
Газы с высокой теплотой сгорания, приближающиеся по этому показателю к природному газу, в настоящее время в промышленных масштабах пока не производят. Однако технология их получения в ряде случаев отработана на достаточно крупных опытно-промышленных установках. Основа повышения теплоты сгорания газа — обогащение его метаном за счет проведения газификации при повышенном давлении, благодаря чему интенсифицируется взаимодействие углерода и его оксидов с водородом, образующимся в слое топлива. Продуктом этих реакций является метан. [7]
Разработано также несколько вариантов многоступенчатых газогенераторов, в которых предусмотрены максимальное извлечение летучих продуктов из топлива и последующая газификация углеродного остатка с применением водородсодержащих газов в качестве газифицирующего агента (гидрогазификация). Наряду с этим газ, обогащенный метаном, может быть получен из низко- и среднекалорийного газа путем гидрирования содержащихся в нем оксидов углерода в выносном реакторе (вне газогенератора).
Для современной химической промышленности и энергетики требуются газогенераторы с единичной мощностью по углю 100 т/ч и более. К началу 1970-х годов в промышленном масштабе было реализовано три типа газогенераторов [6].
· слоевые газогенераторы. В разное время действовало более 800 газогенераторов, в том числе более 30 газогенераторов “Лурги” с единичной мощностью по углю до 45 т/ч. После 1977 г. введено в эксплуатацию еще 130 газогенераторов “Лурги”.
· газогенераторы Винклера с кипящим слоем. Было сооружено более 40 аппаратов с единичной мощностью до 35 т/ч по углю.
· пылеугольные газогенераторы Копперса-Тотцека. К началу 1970-х годов эксплуатировалось более 50 аппаратов с единичной мощностью до 28 т/час по углю.
Не случайно все самые мощные газогенераторы имели немецкое происхождение. Причина в том, что в Германии нет собственной нефти, но имеются большие запасы угля. В 1920-1940 гг. в Германии была реализована беспрецедентная по масштабам программа углепереработки с производством моторных топлив, металлургического топлива, газов различного назначения и широкого спектра продуктов углехимии, включая пищевые продукты. Во время второй мировой войны с использованием жидких продуктов пиролиза, прямого и непрямого ожижения угля производилось до 5,5 млн. т в год моторного топлива. Именно немецкие разработки того времени определили на многие десятилетия стратегию развития технологий углепереработки, в том числе газификации топлива.
Если проанализировать конструктивные особенности и принцип действия современных промышленных газогенераторов (к настоящему времени до промышленного масштаба доведено еще более десяти конструкций газогенераторов), можно выделить четыре основополагающих инженерных решения.
1. Создание Фрицем Винклером (концерн BASF) в 1926 г. газогенератора с кипящим слоем. Эта технология послужила основой для современных процессов HTW (Hoch-Temperatur Winkler) и KRW (Kellogg-Rust-Westinghouse) и др.
2. Разработка фирмой "Лурги" в 1932 г. слоевого газогенератора, работающего под давлением 3 МПа. Использование повышенного давления для интенсификации процесса газификации реализовано почти во всех современных промышленных газогенераторах.
3. Разработка Генрихом Копперсом и Фридрихом Тотцеком в 1944-45 гг. пылеугольного газогенератора с жидким шлакоудалением. Первый промышленный аппарат этого типа был построен в 1952 г. в Финляндии. Пылеугольный принцип газификации с жидким шлакоудалением реализован в промышленных аппаратах Destec, Shell, Prenflo, разработанных на основе газогенератора Копперса-Тотцека, в аппарате Texaco и др. Удаление шлака в жидком виде реализовано в слоевом газогенераторе BGL (British Gas– Lurgy), разработанном на основе газогенератора Лурги.
4. Разработка фирмой Texaco в 1950-е годы газификаторов для переработки тяжелых нефтяных остатков. Всего построено более 160 таких установок. В 1970-е годы была разработана модификация аппарата Texaco для газификации водо-угольной суспензии. Принцип подачи угля в аппарат в виде водо-угольной суспензии использован и в газогенераторе Destec.
Были попытки использовать и ряд других технических решений для создания новых газогенераторов: использование внешнего теплоносителя, в том числе тепла ядерного реактора; газификация в расплавах солей, железа, шлака; двух - трехступенчатая газификация; газификация в плазме; каталитическая газификация и др., но они не привели к созданию современного конкурентоспособного технологического процесса.
4. Конверсия метана в синтез-газ
Углекислотная конверсия метана в синтез-газ СО + Н2 - одна из важнейших химических реакций, пригодная для промышленного получения водорода и дающая начало синтезу углеводородов (жидкое топливо) и других технически ценных продуктов.
Существует три метода окислительной конверсии метана в синтез-газ:
· паровая конверсия
CH4 + H2O ↔ CO + 3H2
ΔН = +206 кДж/моль (1)
· парциальное окисление кислородом
CH4 + 1/2O2 ↔ CO + 2H2
ΔН = -35,6 кДж/моль (2)
· углекислотная конверсия
2CO + 2H2 ↔ CH4 + CO2
ΔН = +247 кДж/моль (3)
В промышленности используется практически лишь метод паровой конверсии (1). Реакцию проводят на нанесенном Ni-катализаторе при высокой температуре (700 - 900 °С). Что касается реакции (2), то на ее основе фирмой «Shell» был разработан технологический процесс в некаталитическом варианте при очень высоких температурах (1100 - 1300 °С), реализованный на небольшом заводе в Малайзии. Заметим, что по последним сведениям из-за аварии этот завод сейчас не работает. Реакция (3) пока находится в стадии исследования на уровне лабораторных и пилотных испытаний. Как следует из уравнений (1) - (3), количественный состав образующегося синтез-газа в этих реакциях различный: в реакции (1) получается синтез-газ состава СО:Н2 = 1:3, в реакции (2) - смесь 1:2, в реакции (3) - смесь 1:1. Потребность в синтез-газе того или иного состава определяется его последующим техническим назначением.
Так, для синтеза метанола требуется синтез-газ состава 1:2
СО + 2Н2 = СН3ОН (4)
В производстве аммиака из азото-водородной смеси на стадии ее получения применяют синтез-газ состава 1CO:3H2. Относительно недавно предложено использовать синтез-газ состава 1:1 для промышленного получения диметилового эфира [9, 10]. Формальная стехиометрия этой реакции соответствует уравнению
2СО + 4Н2 = СН3ОСН3 + Н2О (5)
Однако, с учетом того, что в условиях этого процесса H2O вступает во взаимодействие с CO (паровая конверсия CO)
CO + H2O ↔ CO2 + H2
ΔH = - 41 кДж/моль (6)
реально для получения диметилового эфира требуется смесь CO:H2 состава 1:1:
3СО + 3Н2 = СН3ОСН3 + СО2 (7)
Термодинамическое рассмотрение реакции (7) указывает, что она может осуществляться при давлениях значительно меньших, чем реакция (4). Катализатором реакции (7) может служить комбинация катализаторов дегидратации и синтеза метанола. Получаемый диметиловый эфир предлагается применять в качестве топлива в дизельных двигателях без переделки самих двигателей (это топливо резко снижает вредные выхлопы ─ «топливо 21 века», как его назвали разработчики).
4.1 Термодинамика процесса
Равновесие в системе 2CO + 2H2 ↔ CH4 + CO2
Большие трудности в практическом осуществлении всех методов конверсии метана связаны со значительным тепловым эффектом: как эндотермичность реакций (1) и (3), так и экзотермичность реакции (2) создают проблему подвода или отвода тепла. В углекислотной конверсии метана (3) при 700 - 800 °С на многих никелевых и платиновых катализаторах достигается равновесная конверсия в синтез-газ СО + Н2. В этих условиях одновременно с реакцией (3) осуществляется взаимодействие монооксида углерода с водяным паром (6). Протекание реакции (6) приводит к тому, что в равновесии (3) отношение СО:Н2 оказывается меньше 1, а конверсия СО2 больше конверсии СН4. Лишь при 900 °С и атмосферном давлении выход Н2 и СО приближается к 100%, а отношение Н2О/СО к нулю.
На рис. 11 показана зависимость равновесного выхода Н2 и СО в исходной системе CH4 + CO2 от температуры и давления.
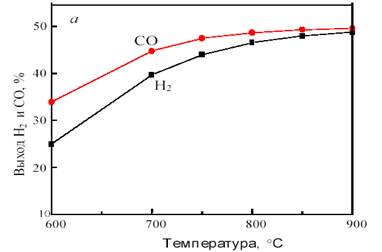

Рис. 11. Зависимость равновесного выхода Н2 и СО от температуры при 0,1 МПа (а) и от давления при 800 °С (б) в исходной смеси 1СН4:1СО2
Как видно из рис. 11, с повышением температуры выход водорода и CO возрастает, достигая предела вблизи 900 °С. С ростом давления равновесная конверсия уменьшается.
Основным препятствием к использованию Ni-катализаторов отравляемость коксом. Возможны два пути образования кокса при разложении метана:
· диссоциация метана
СН4 = С + 2Н2
ΔН = +74,8 кДж/моль•С (8)
· реакция Будуара
2CO ↔ C + CO2
ΔН = -172,5 кДж/моль•С (9)
Первая из них ─ эндотермическая, вторая ─ экзотермическая. Обе реакции могут быть представлены как стадии суммарной реакции (3). Однако в реальности они протекают при разных температурах: реакция (8) ─ преимущественно при высоких температурах, реакция (9) ─ при низких температурах, и в реальных условиях кокс почти всегда образуется. Согласно термодинамическим соображениям суммарное углеотложение должно снижаться с повышением температуры. Действительно, эксперимент подтверждает, что основное количество углерода образуется по реакции (8), а не (9). Часто углерод, диффундируя в металл, образуется на выходе из катализатора в виде нитей.
Одним из путей решения проблемы, связанной с подводом и отводом тепла при получении синтез-газа, является разработка процесса комбинированной конверсии смеси СН4 + СО2 + Н2О + О2, в котором бы без дополнительного подогрева сочетались реакции (1), (2), (3) и (6). Такую термонейтральную (автотермическую) конверсию можно осуществить, комбинируя углекислотную (3) и кислородную (2) конверсию метана в системе СН4 + СО2 + О2. Термодинамический расчет процесса комбинированной конверсии, включающей реакции (2), (3) и паровой конверсии СО (6), показывает, что в смеси 50% СН4 + (50 - х)% СО2 + х% О2 при 800 °С термонейтральность достигается при х = 23% (рис. 12). В реакции смеси 50% СН4 + 27% СО2 + 23% О2 при 800 °С и 1 атм. равновесные выходы составляют: 49,3% Н2 и 36,5% СО, т.е. соотношение CO:H2 сильно отличается от единицы.
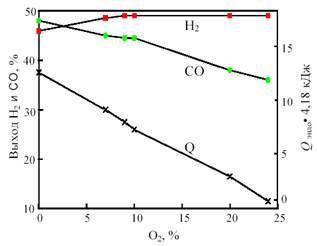
Рис. 12. Зависимость равновесного выхода Н2 и СО и теплового эффекта реакции (теплопоглощения) Qэндо при 800 °С и 0,1 МПа от содержания кислорода (x) в смеси 50% СН4 + (50-х)% СО2 + х% О2
Изменение соотношения исходных компонентов позволяет получить газ состава 1СО:1Н2 с одновременным сохранением термонейтральности. Например, исходная смесь, содержащая 38% СН4, 43% СО2 и 19% СО2, при 800 °С и 1 атм. дает продукт состава 36,0% Н2 и 36,4% СО при нулевом тепловом эффекте. При повышении температуры получается избыток СО: при 900 °С ─ 34,6% Н2 и 38,0% СО, а при снижении температуры ─ избыток Н2: при 700 °С ─ 36,4% Н2 и 33,6% СО. В качестве примера на рис. 13 показана зависимость равновесного выхода СО и Н2 от температуры и давления для исходной смеси 38 % CH4 + 43% CO2 + 19% O2.
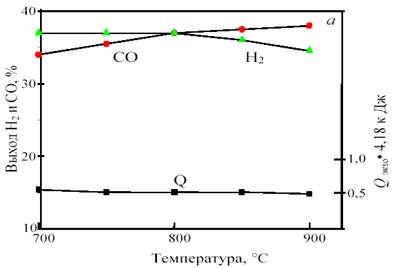
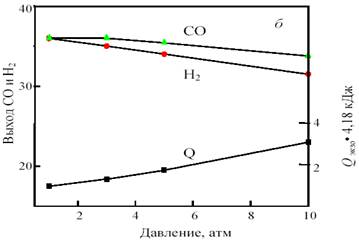
Рис. 13. Зависимость равновесного выхода Н2 и СО и теплового эффекта реакции (тепловыделения) Qэндо при 0,1 МПа от температуры (а) и при 800 °С от давления (б) в смеси 38%СН4 +43%CO2 + 19%O2
Важно отметить, что для этой смеси, в отличие от смеси 1СО:1Н2, с ростом давления от 1 до 10 атм. равновесный выход продуктов уменьшается не намного, всего на 2─3%. Это позволяет интенсифицировать процесс путем увеличения давления без изменения соотношения продуктов и термонейтральности.
4.2 Кинетика углекислотной конверсии метана
Первой работой по кинетике углекислотной конверсии метана (3) была работа, выполненная в лаборатории М.И. Темкина [11]. Основываясь на схеме

они показали, что в случае протекания процесса на никелевой фольге при 800─900 °С реакция описывается таким же кинетическим уравнением, что и паровая конверсия (1) на этом же катализаторе [27]:
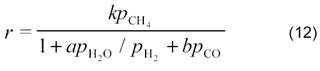
где k, a и b ─ константы; ρСН4, ρН2О, ρН2 и ρСО ─ парциальные давления метана, воды, водорода и СО, соответственно.
Если в смеси имеется водяной пар, то фактически протекает паровая конверсия СН4 с повторным быстрым образованием воды по реакции, обратной (6). В работе [11] подтверждается, что конверсия смесей СН4+СО2 и СН4+Н2О на катализаторе Ni/МgО соответствует одинаковому кинетическому уравнению.
В дальнейшем были найдены и другие уравнения. Например, для реакции, соединяющей углекислотную конверсию метана (3) и паровую конверсию СО (6):
СН4 + 2СО2 → Н2 + Н2О + 3СО (13)
на Ni/C, Ni/SiO2, Ni/TiO2 и Ni/MgO, а также на нанесенных Pt-катализаторах [12] было получено кинетическое уравнение:
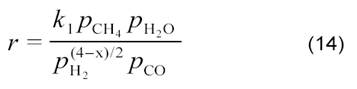
По мнению авторов [12], оно соответствует схеме:
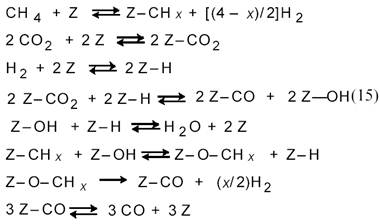
Для процесса на катализаторе Ni/Al2O3 было получено уравнение:
r = kp1/2 (16)
Существует сводка данных (взятых из более 60 статей) по исследованию кинетики углекислотной конверсии метана. Ниже дана краткая оценка этих данных.
Значения энергии активации Еа изменяются в интервале:
· по расходованию CH4 и CO2 соответственно от 30 до 350 кДж/моль и от 35 до 130 кДж/моль,
· по выходу CO и H2 соответственно от 38 до 218 кДж/моль и от 38 до 250 кДж/моль.
Наиболее выпадающие значения ЕСН4 относятся к реакции на Re/Al2O3 350 кДж/моль) и Pt (73 кДж/моль). Некоторые исследователи считают, что из числа достоверных данных следует также исключить значения энергии активации для реакции на Pb/MgO, т.к. EСО > EН2, причем ЕСО = 921 кДж/моль. Большинство остальных данных для Ni-катализаторов находятся вблизи значения 239 ± 20 кДж/моль, которое довольно близко к энергии активации диссоциации СН4 на Ni(110) и Ni(111): 233 ± 27 и 221 ± 20 кДж/моль, соответственно. Для катализа с участием благородных металлов Еа ближе к 314─377 кДж/моль.
На кажущиеся значения энергии активации углекислотной конверсии метана сильно влияет реакция (6), что отражается, по-видимому, в увеличении ЕСН4 с ростом объемной скорости. При этом снижается конверсия и влияние обратной реакции (гидрирование СО в СН4)становится менее значительным.
Каталитическая активность при 450 °С (экстраполяция), выраженная через число оборотов реакции tn, изменяется в интервале от 0,1 до 1,0.
Большие значения tn получены для Ru/TiO2 (4,3.7,2), Ru/Al2O3 (1,5─4,3), Rh/VOx/SiO2 [4]. Для кристалликов Rh на носителе получен следующий ряд tn: ZrO2 > TiO2 ≥ Al2O3 > La2O3 = SiO2 > MgO. Этот ряд совпадает с рядом: TiO2 > Al2O3 > SiO2 и не совпадает с рядами: Al2O3 > La2O3 > CeO2 > MgO > TiO2 и MgO > TiO2 ≈ Al2O3 > SiO2 [13].
Было установлено, что число оборотов реакции не зависит от природы носителя: ZrO2 ≈ TiO2 ≈ Al2O3 ≈ SiO2. Такие противоречия могут быть объяснены влиянием обратной реакции, измерениями при разных объемных скоростях или неправильностью экстраполяций. Для нанесенных Ni-катализаторов получен следующий ряд tn:TiO2 > Al2O3 ≈ SiO2 ≈ MgO.
В подавляющем числе исследований установлено, что скорость углекислотной конверсии метана пропорциональна давлению СН4 в первой степени, в то время как величина ρCOn входит в кинетические уравнения, приведенные в разных работах, в числитель и знаменатель с показателем степени n от 0 до 2. Это указывает на то, что взаимодействие метана с катализатором является лимитирующей стадией.
Константы скорости взаимодействия СН4 и СО2 с единичным Ni-центром на Ni/TiO2 были измерены при 420 °С. С повышением температуры восстановления катализатора способность к диссоциации СН4 растет, а диссоциации СО2 не изменяется.
Положительный кинетический изотопный эффект (КИЭ) kCH4/kCD4, наблюдавшийся в процессе на Ni/γ-Al2O3, Ni/SiO2, Rh/SiO2 , Ni/La2O3 [13], также указывает на то, что стадия активации метана является лимитирующей, а диссоциация СО2 происходит легко. Для реакций на Ni/Al2O3 и Ni/La2O3 величина КИЭ растет с повышением температуры, причем в случае Ni/La2O3 КИЭ значительно выше, чем в процессе на Ni/Al2O3.
Исследование кинетики углекислотной конверсии метана на Ni/SiO2 при 700 0С и атмосферном давлении [13] позволило получить следующие данные: реакция первого порядка по ρСО2 и по ρН2, с ростом ρСН4 скорость реакции быстро увеличивается и достигает насыщения.
Для описания кинетики предложена схема

Лимитирующей стадией является поверхностное взаимодействие адсорбированных углерода и кислорода. По данным [13], кинетика углекислотной конверсии метана сильно зависит от обратной реакции ─ гидрирования CO:
![]()
Энергия активации реакции по расходованию метана (ECH4) растет в ряду Ru/TiO2, Ru/Al2O3, Ru/C: 76,4; 107,4; 107,6 кДж/моль, соответственно. Такая же закономерность наблюдается для ECO2: 71,6; 75,4; 86,2 кДж/моль, что отвечает эффекту сильного взаимодействия металл-носитель.
EH2 = 17,1; 18,0; 20,6 кДж/моль, соответственно, была всегда больше ECO: 97,1; 125,2; 111,3 кДж/моль.
Для реакции на сульфидных катализаторах MoS2 и WS2 при 600 °С получено следующее кинетическое уравнение:
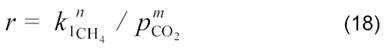
которое отличается от уравнений (14, 16, 17) для реакции на нанесенных металлических катализаторах. Расхождения объясняются большой адсорбцией СО2 и малой адсорбцией СН4. Наличие СО2 на поверхности подавляет разложение метана.
Для реакции на оксидно-марганцевых катализаторах в наших работах [14] было получено кинетическое уравнение
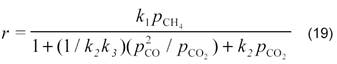
В случае малых конверсий уравнение имеет более простой вид:
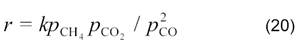
Практически все исследователи отмечают, что конверсия СО водяным паром протекает с большими скоростями, чем углекислотная конверсия СО2.
4.3 Механизм конверсии смеси CH4 + CO2
В большинстве предлагаемых механизмов углекислотной конверсии метана рассматривается диссоциативная адсорбция метана и СО отличающаяся от схемы (11) отсутствием стадии взаимодействия СНх с водой [15]. Предполагается последовательная диссоциация СН4 на поверхности образованием частиц СНх и С и их взаимодействие адсорбированным атомом О, а не с водой. Эти процессы отражает схема:
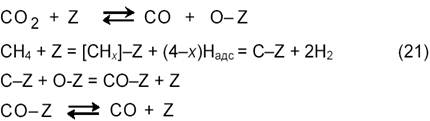
Диоксид углерода может также непосредственно реагировать с поверхностным углеродом по реакции, обратной реакции Будуара (9). По данным [18] диссоциация метана на никеле протекает преимущественно на малых кристалликах металла. Обнаружен также эффект структурной чувствительности диссоциации метана, на гранях кристалла Ni диссоциация CH4 следует ряду: Ni(110) > Ni(100) > Ni(111). Как показано импульсным методом, в условиях реакции (3) в зависимости от природы катализатора образуются различные промежуточные соединения CHx: х = 2,7 для Ni/MgO, 2,5 для Ni/SiO2, 2,4 для Ni/Al2O3, 1,9 для Ni/TiO2, 1,0 для Со/SiO2 и 0,75 для Co/Al2O3. Вещества СНх с малыми значениям х легче дают углеродные отложения. По мнению, Н-спилловер на носитель минимизирует углеобразование, сдвигая равновесие в сторону CHx с большими значениями х. Скорость разложения СН4 в условиях реакции (3), по-видимому, выше, чем скорость разложения одного СН4, без участия CO2. Поэтому схема (11) нуждается в соответствующих уточнениях.
С механизмом (21) согласуется ряд экспериментальных данных. Так, величина КИЭ для конверсии смеси CH4 + CO2 на Ni/SiO2 больше, чем в случае образования СО только из метана. Это объясняется двумя маршрутами генерации СО: одна молекула СО образуется из СН4, а другая ─ из СО2 [11]:

Подобный же результат был получен с помощью метода изотопного обмена и ИК-спектроскопии диффузного рассеяния.[16] Диссоциативная адсорбция метана и СО2 на нанесенном родиевом катализаторе была доказана в экспериментах с мечеными молекулами 13СН4 и С18О2. Таким образом, на основании проведенных исследований можно заключить, что реакция (3) протекает по окислительно-восста-новительному механизму (21): СО2 окисляет поверхность катализатора, а СН4 восстанавливает ее.
Большой цикл работ по исследованию механизма углекислотной конверсии метана на Ni/SiO2, Ni/La2O3, Ru/SiO2, Ru/Al2O3 и других катализаторах методами ТАР-реактора (TAP ─ temporary analysis of products), изотопного обмена, рентгеновской фотоэлектронной спектроскопии, ферромагнитного резонанса, электронной микроскопии и ИК-спектроскопии выполнен К. Миродатосом с сотр. [16─17]. Исследование конверсии CH4 на Ni/SiO2 методом изотопного обмена (12СН4 и 13СО2) показало, что после импульса 12СН4 наблюдается быстрое выделение Н2, а на поверхности катализатора, очевидно, остается слой карбида Nix12C, наиболее вероятный его состав Ni2C или Ni3C. Карбидоподобные формы в условиях реакции остаются стационарными и могут гидрироваться обратно в СН4. После импульса 13СО2 регистрируются два импульса СО: сначала образуется 13СО из 13CO2, на поверхности остается Оадс, затем образуется 12СО за счет взаимодействия Оадс с СН4 или с 12Садс после разложения СН4. Решеточный подповерхностный кислород в образовании CO и H2 не участвует. Диоксид CO2 находится в обратимом равновесии с поверхностью и с первой молекулой СО. Образование второй молекулы СО является лимитирующей стадией и лимитирует здесь медленная диффузия атомов С и О. Таким образом, согласно [16─17] и вопреки мнению большинства других исследователей, в углекислотной конверсии метана на Ni/SiO2 лимитирующая стадия не включает диссоциацию С─Н-связи, а небольшое значение КИЭ (kCH4/kCD4) может быть объяснено разрывом С─Н-связи в обратимой стадии диссоциации метана.
В отличие от реакции на Ni/SiO2, для процессов на Ru/SiO2, Ru/Al2O3, Ru/C лимитирующей стадией является диссоциация СН4, а затем СО2 реагирует с адсорбированным углеродом с образованием СО. Накопление углерода здесь минимально и, следовательно, выделение водорода и последующее его окисление подавлено.
На катализаторе Ru/SiO2, поскольку SiO2 является довольно инертным носителем, вся реакция CH4 + CO2 протекает на фазе Ru. Быстрое отравление катализатора вызвано образованием промежуточного
углерода, склонного к полимеризации и дальнейшей графитизации. В случае реакции на Ru/C носитель-графит собирает частички CHx, что уменьшает время жизни образующегося углерода на Ru и обусловливает
очень высокую стабильность этого катализатора. В реакции на Ru/Al2O3 участвуют также группы AlOH, подпитываемые спилловером адсорбированных частиц H и O с Ru, что ограничивает дезактивацию катализатора.
В общем случае на Ru-фазе нанесенных рутениевых катализаторов протекают необратимая диссоциация CH4 и следующие процессы:
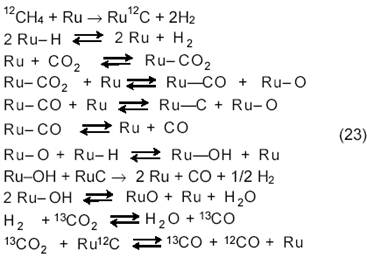
Последнее уравнение показывает, что обе молекулы СО образуются в одной реакции на катализаторе Ru/SiO2, но имеют разное происхождение.
На Ru/Al2O3 протекают еще реакции с участием AlOH-групп:
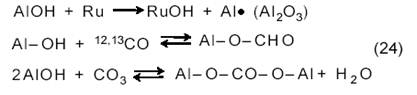
К выводу о простом механизме со стехиометрическим разложением СН4 на основании импульсных измерений приходят также в работах [13, 14]. Однако здесь следует иметь в виду, что импульсный метод может и не выявить образования частиц СНх, которые принимаются большинством авторов как промежуточные. Есть данные [15], что на нанесенных Ni-катализаторах число оборотов для разложения СН4 на С и Н2 много ниже, чем для конверсии СН4 с СО2.
Более сложная картина наблюдается в случае реакции на Ni/La2O3. Как показывает метод изотопного обмена, молекулы 13СО и 12СО получаются при взаимодействии СН4 и СО2 с катализатором:
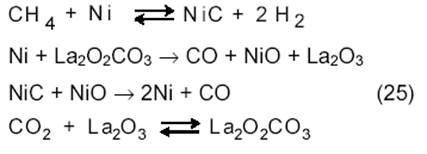
На металлах, нанесенных на оксид с основными свойствами, реакция протекает на границе металл-оксид, причем метан диссоциирует на металлической поверхности, а СО2 образует карбонат на носителе. Таким образом, катализатор Ni/SiO2 является монофунциональным, а катализатор Ni/La2O3 ─ бифункциональным.
Методы ТАР-реактора и ИК-спектроскопии показали, что интенсивность линий СО2 после впуска смеси СО2 + СН4 на Rh/γ-Al2O3 проходит через максимум, а затем образуется СО32─, так что механизм с участием реакции Будуара весьма вероятен.
Методы ТАР-реактора и ИК-спектроскопии применили также для исследования конверсии смеси СН4+СО2 на катализаторах ZrO2 и Pt/ZrO2. Установлено, что селективность образования СО определяется одним и тем же промежуточным веществом и зависит от содержания атомарного кислорода на каталитической поверхности. Оксид ZrO2 ответственен за активацию СО2, он частично восстанавливается и реокисляется в условиях реакции. Замещение решеточного кислорода в ZrO2 кислородом из СО2 ─ медленная стадия процесса. После импульса СО2 единственными регистрируемыми частицами, которые остаются достаточно долго на поверхности, являются поверхностные ОН-группы. Очевидно, Оадс после активации СО2 реагирует с метаном. Природа Оадс неясна, это могут быть и реакционноспособные ОН- или СО3-группы. Метан не диссоциирует на ZrO2, но диссоциирует на Pt/ZrO2.
Катализатор Pt/ZrO2 активнее, чем платина на других носителях. Возможно, при этом образуется сплав Pt1─xZr. Предложена следующая схема механизма конверсии на Pt/ZrO2:
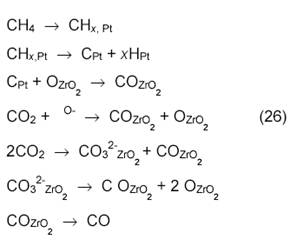
Еще в одной работе с использованием метода ТАР-реактора [18] показано, что не атомы О, а поверхностные ОН-группы реагируют с поверхностными частицами С или СНх:
![]()
Однако в большинстве работ ключевыми промежуточными частицами считают адсорбированные атомы кислорода. В работе [13] предполагается корреляция скорости образования СО в смешанной конверсии СН4+СО2+Н2О с прочностью связи металла с Оадс. Такую корреляцию нельзя считать достоверной и даже если она существует, это еще не доказывает, что атомы Оадс являются ключевыми интермедиатами.
Исследование реакции (3) на NiO/MgO при 800 °С (изотопный метод) показало, что в этих условиях на катализаторе присутствуют два типа кислорода: адсорбированный, взаимодействующий с Садс, и решеточный, реагирующий значительно медленнее. Реакция (3) протекает по окислительно-восстановительному механизму.
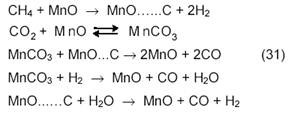
Окислительно-восстановительный механизм углекислотной конверсии метана на Ni-катализаторах, нанесенных на MgO, принимается и в работах Института химической физики РАН. Окисление Ni и восстановление NiO ускоряется на катализаторе Ni-Cr2O3/MgO. Процесс протекает через промежуточное образование шпинели по суммарному стехиометрическому уравнению:

Диссоциация метана происходит, по-видимому, на Ni или NiC, а активация CO2 осуществляется за счет участия кислорода шпинели NiCr2O4, вероятно, через промежуточное образование карбоната MgCO3.
Рассмотрена возможность окислительно-восстановительного механизма углекислотной конверсии метана на системе Pt/CeO2. Здесь Pt активирует CH4, а CeO2─x активирует (восстанавливает) CO.
В ряде работ фиксировали образование карбонатов на металлических катализаторах, нанесенных на основные носители. Например, было показано [13], что на Pt/ZrO2 восстановление СО2 происходит через образование карбоната циркония вблизи границы ZrO2 с Pt. Углерод на металле восстанавливает этот карбонат до формиата.
Далее протекают реакции:
Можно предположить, что на разных катализаторах механизм различен: на чистых металлах и на металлах, нанесенных на нейтральные носители типа SiO2, более вероятна полная диссоциация СН4 и СО2, а на металлических катализаторах с основными носителями вероятнее промежуточное образование карбоната.
В ИК-спектрах поверхностных соединений, а также после завершения реакции (3), были обнаружены монодентатные и бидентатные комплексные карбонаты, гидрокарбонаты, формиатные комплексы, линейные и мостиковые карбонилы, группы СНх и НСО. Предполагается участие в некоторых механизмах и этих промежуточных веществ. Показано, что атомы Н способствуют разложению карбонатов.
В условиях углекислотной конверсии метана на Pt/TiO2, Pt/SiO2, Pt/ZrO2, Pt/Cr2O3 преобладающим поверхностным соединением является адсорбированный СО. Обнаружен также СН2О. По мнению авторов [12], важнейшим интермедиатом на поверхности является СНхО., хотя прямых ИК-спектроскопических подтверждений пока нет. Тем не менее авторы [12,13] считают, что полученные ими данные свидетельствуют в пользу механизма (15) с вероятными стадиями: обратимая диссоциация СН4 с образованием СНх и Н2, недиссоциативная адсорбция СО2 на носителе, диссоциация адсорбированного СО2 с участием Н на границе металл-носитель, реакция СНх с О (или с ОН) на границе металл-носитель с образованием СНхО. и последующее разложение этого интермедиата с образованием продуктов реакции СО и Н2.
По ИК-спектроскопическим данным других исследователей [14] на катализаторах Pt/TiO2 и Pt/ZrO2 адсорбция и активация СО2 протекают по механизму обратной конверсии водяного газа (6) на носителе с участием поверхностных ОН-групп. Образуются группы СНхО, а при их разложении ─ СО и Н2.
Механизм начальных стадий конверсии СН4+СО2 на никеле, нанесенном на SiO2, La2O3•SiO2 или La2O3, изучали импульсным методом отклика [18]. На NiO адсорбция СО2 конкурирует с диссоциацией СН4. На La2O3 диоксид углерода адсорбируется с образованием карбонатов и формиатов, которые затем разлагаются с выделением СО и восстановлением кислородных вакансий. Ресурсы О для образования СО из СН4 обеспечиваются за счет перехода кислорода от La2O3 к Ni. Метан восстанавливает NiO и образует вакансии в La2O3.
По данным [10], в случае катализа на NiMgO, промотированном Pt, Pd или Rh, лимитирующей стадией реакции (3) становится диссоциация СО2 или поверхностная реакция СНх + Оадс вместо диссоциации СН4 (для непромотированного NiMgO).
Есть также предположения о промежуточном образовании метильных радикалов. Для выяснения этого механизма на примере конверсии СН4+СО2 на Rh/SiO2 на катализатор адсорбировали радикалы СН3, полученные разложением азометана [18]. По ИК-спектрам было установлено, что частицы СН3,адс реагируют с О2 и СО2 из газовой фазы при температуре более 100 °C, поэтому кокс не отлагается на катализаторе. При этой же температуре радикалы СН3,адс разлагаются в вакууме, причем разложение ускоряется под действием СО2.
Большая часть СН3 адсорбируется на носителе SiO2, а Rh участвует в дальнейших превращениях. Метан при этой температуре на катализаторе не адсорбируется.
Предполагается протекание следующих реакций:

Существование СН3-групп в условиях реакции СН4+СО2 на Ni/SiO2 показано также методами температурно-программируемого восстановления и температурно-программированной реакции (ТПР) [18]. Наблюдалось образование С2Н6. Группы СН3 на поверхности могут взаимодействовать с адсорбированными атомами О с образованием групп СНхО. и далее СО и Н2. Проводилось изучение механизма углеобразования. Установлено, что после диссоциации СО на Ni атомы углерода мигрируют в подповерхностный слой никеля, индуцируя его реконструкцию, удлинение связей Ni─Ni и последующее более глубокое проникновение в металлический кристаллит вплоть до отложения углерода на обратной поверхности кристаллита. Электронно-микроскопическое исследование показало разные свойства углерода, образовавшегося по реакциям (8) и (9). В случае смеси СО+СО2 углерод капсулируется, а из смеси СН4+Н2 формируются графитовые пластинки и нити. Отсюда следует вывод, что вначале образуется С из СО/СО2, а вторичный углерод осаждается при диссоциации метана.
Методом температурно-программированного гидрирования после завершения реакции на Ni/MgO были обнаружены две формы углерода: аморфный α-С, гидрирующийся при 270─420 °С и β-С, гидрирующийся выше 600 °С, по-видимому, это графит. Углерод образуется преимущественно на малых частицах никеля.
Проведение температурно-программированного процесса взаимодействия СО и СН4 на катализаторах Ni0,03Mg0,97O, 3%Ni/MgO и 3%Ni/Al2O3 позволило выявить, что углерод образуется как при диспропорционировании СО, так и при диссоциации метана [18]. При
этом происходит быстрое окисление СНх на Ni под действием СО2. На всех трех изученных катализаторах температура пика взаимодействия С+СО2 одна и та же ─ 550 °С), что указывает на отсутствие взаимодействия между катализатором и осажденным углеродом. На восстановленном катализаторе Ni0,03Mg0,97O реакция между СО2 и Ni протекает при температуре на 40 °С ниже, чем на остальных катализаторах. Авторы [18]
приходят к выводу, что возможны два маршрута активации СО2:
1) на носителе вблизи границы с Ni и 2) на частицах Ni.
Первый маршрут более благоприятен для ингибирования углеобразования.
В опытах со смесями 13СН4 + 12СО2 на катализаторе Ni/CaO-Al2O3 также было установлено, что углерод образуется как из СО, так и из СН4 [38]. Высокотемпературный пик поглощения Н2 в ТПР-экспериментах (450─600 °С) приписан спилловеру водорода с Ni на носитель.
Квантово-механический расчет реакции СО2+СН4 на Cu(111), Ni(111), Pd(111), Pt(111), Rh(111), Ru(111), Ir(111) и Fe(111) методом UBI─QEP (unity bond index ─ quantum exponential potential) [18] показал, что лимитирующими стадиями являются как диссоциация СН4, так и диссоциация СО2, причем оба процесса ускоряют друг друга. Рассмотрев 84 возможных реакции на поверхности, авторы пришли к следующей более вероятной схеме:
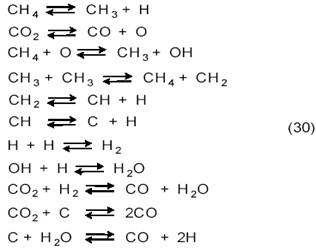
Ряд каталитической активности, по их расчетам, примерно соответствует экспериментальным данным:
Fe > Ni > Rh > Ru > Ir > Pd > Pt > Cu.
Однако для практики Ni предпочтительнее Fe, потому что никель менее подвержен коксоотложению, а Ru лучше Rh, поскольку рутений дешевле.
Промежуточное образование карбонатов в углекислотной конверсии метана на оксидных катализаторах было доказано в наших работах [14]. Согласно кинетическим данным и методу термодесорбции и рентгенофазового анализа на нанесенных оксидно-марганцевых катализаторах процесс протекает по механизму
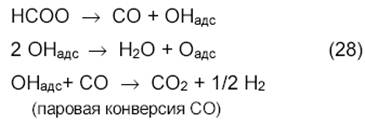
Таким образом, продукт реакции ─ СО образуется в результате восстановительного разложения карбоната при его взаимодействии с углеродом (или карбидом, или СНх) или с Н2.
До сих пор не был рассмотрен вопрос о возможных гомогенных стадиях в углекислотной конверсии метана. Между тем, появились сведения о гетерогенно-гомогенном механизме другой эндотермической реакции, а именно, паровой конверсии СН4 в синтез-газ (1). Авторы [19, 20] связали наблюдаемое ими увеличение скорости реакции при уменьшении навески Ni-катализатора ГИАП-16 с реализацией гетерогенно-гомогенного механизма с вылетом в объем промежуточных частиц, например метильных радикалов. Кинетика и механизм реакций (1) и (3) близки. Поэтому не исключено наличие гомогенных стадий и в конверсии СН4+СО2. Также был обнаружен аналогичный эффект для реакции (3) ─ небольшой рост конверсии с уменьшением навески Ni-Cr/MgO-катализатора. Однако величина эффекта не настолько велика, чтобы принять его как доказательство гетерогенно-гомогенного механизма. Таким образом, совокупность данных большого числа работ доказывает существование двух основных механизмов реакции СН4+СО2:
1) диссоциация СН4 и СО2 и последующее взаимодействие атомарных фрагментов С, Н и О на массивных металлических катализаторах или на металлах, нанесенных на инертные носители, и
2) диссоциация СН4 и взаимодействие фрагментов С или СHx с СО2 или с СО32─ на металлах, нанесенных на основные носители. В последнем случае взаимодействие облегчается в присутствии добавок оксидов переходных металлов, участвующих в восстановлении CO2.
4.4 Катализаторы углекислотной конверсии метана
Нанесенные никелевые катализаторы.
Наибольшую активность в углекислотной конверсии СН4 проявляют нанесенные никелевые катализаторы. Однако они имеют существенный ─ потеря активности при закоксовывании. Для борьбы с этим явлением применяются разные приемы. Так, в процессе SPARG, разработанном фирмой «Topsoe», углеотложение на никеле подавляется путем пассивации серой. Считают, что сера препятствует образованию больших ансамблей углерода и таким образом ингибирует процесс углеотложения сильнее, чем реакцию (3).
Наименее подвержены влиянию кокса катализаторы, в которых никель нанесен на основные носители. Так, если катализатор Ni/Al2O3 обладает наибольшей активностью в начальный период работы, то катализаторы Ni/MgO, Ni/CaO, Ni/MnO, Ni/ZrO2 превосходят его по эксплуатационным качествам, проявляя устойчивость в отношении коксообразования. Отмечается [10], что углеотложение подавляется, если металл нанесен на носитель с высокой основностью по Льюису. На таких оксидах, как СaO, MgO, TiO2, адсорбированный диоксид углерода реагирует с углеродом по реакции, соответствующей обратной реакции Будуара (8):
![]()
Применяют также щелочные добавки к таким носителям, как Al2O3. По-видимому, образование не слишком стабильных карбонатов облегчает их взаимодействие с углеродом.
Несомненный интерес представляет цикл работ японских исследователей по углекислотной конверсии метана на никелевых катализаторах [11─24]. Методом соосаждения солей Ni и Mg была получена система Ni0,03 Mg0,97O, представляющая собой твердый раствор NiO и MgO, которая оказалась близкой по активности к нанесенному катализатору примерно такого же состава 3%NiO/MgO, но со значительно более высокой коксоустойчивостью. Стабильность обоих катализаторов много выше, чем Ni/SiO2 и Ni/Al2O3.
В условиях низких температур (500 °С) отложение кокса на катализаторе Ni0,03Mg0,97O не наблюдается. При 650 °С активность катализатора не снижается в течение 3000 ч. При более высокой температуре (700─900 °С) на нем также практически не обнаруживается
кокс. В условиях катализа весь никель восстанавливается до металлического состояния, при этом металл выделяется в виде высокодисперсных частиц. Каталитическая активность в конверсии СН4 + СО2 коррелирует с количеством наиболее слабо связанного аморфного α-углерода.
По мнению авторов [11], дезактивация катализатора вызвана не столько углеобразованием, сколько реокислением Ni до NiO. Маленькие частицы Ni, образующиеся в твердом растворе Ni0,03Mg0,97O, восстанавливают СО2 до СО, при этом окисленные частицы NiO в условиях реакции снова восстанавливаются до металлического Ni.
Конверсия смеси СН4 + СО2 на катализаторе Ni0,03Mg0,97O при 850 °С и давлении 0,1─0,2 МПа стабильно составляла 100%, а на катализаторе Ni0,03Ca0,10Mg0,87O ─ 45% [14]. При давлении 1,2 МПа наблюдается углеотложение, которое флуктуирует в ходе работы катализатора. Добавка СаО в этом случае значительно снижает углеобразование (от 330•10─3 г/г катализатора без СаO до 9,5•10─3 г/г катализатора с добавкой СаO).
Промотирование катализатора Ni0,03Mg0,97O благородными металлами (Pt, Pd и Rh) дает максимальный эффект при отношении М : M(Ni + Mg) = 0,021 [15]. На биметаллических катализаторах сильно снижается углеотложение. Кроме того, благородные металлы увеличивают стабильность катализатора при высоких температурах (850 °С).
В [25, 26] показано, что более концентрированные твердые растворы состава NiMgO (13─20% масс. Ni) после восстановления в условиях углекислотной конверсии метана значительно более активные и стабильные, чем системы NiO/Al2O3 и NiO/SiO2 в соответствующей концентрации. При этом лишь часть никеля входит в твердый раствор при реокислении. Вместе с тем смесь NiO + MgO, в отличие от катализатора NiO/MgO, полученного методом пропитки, менее устойчива к спеканию. Из-за взаимодействия никеля с MgO образование кристалликов Ni и, следовательно, отложение углерода уменьшено. Конверсия метана при 790 °С и объемной скорости газового потока 30000 см3/(г•ч) составляет 90%, селективность превращения в СО и Н2 равна 98%.
Различие между нанесенными катализаторами и каталитической системой, представляющей собой химическое соединение между компонентами, выявлено и в случае системы Ni + Al2O3. Катализатор Ni/Al2O3, приготовленный из аэрогеля, более активный и более коксоустойчивый, чем нанесенный катализатор, полученный пропиткой носителя солями Ni [27]. Установлено, что на катализаторе NiAl2O4, сформированном заранее, углеобразование меньше, чем на нанесенном катализаторе Ni/Al2O3, восстановление идет труднее, кристаллики Ni имеют меньшие размеры. Исследование с помощью трансмиссионного электронного микроскопа показало, что на поверхности катализатора образуются углеродные нити. Соли калия увеличивают стабильность катализатора при 650 °С, но при более высокой температуре термостабилизирующий эффект не наблюдается. Согласно [27], фасетированные или плоские частицы металла производят мало нитевидного углерода, а сферические частицы приводят к образованию закапсулированного углерода.
Катализатор 5%Ni/CaO • Al2O3, полученный осаждением никеля на уже сформированный алюминат CaAl2O4, имеет большую активность и менее подвержен отложению углерода, чем катализатор, полученный
смешением солей Ni, Ca и Al. Наблюдаемые различия приписаны разным количествам образовавшегося NiO на каталитической поверхности.
Показано [24], что активный, стабильный и селективный катализатор углекислотной конверсии метана можно получить при нанесении Ni на оксид α-Al2O3, модифицированный путем пропитки раствором Al(NO3)3.
Активность этого катализатора при 650─750 °С в смешанном и углекислотном риформинге ниже, чем в кислородном риформинге. Изучение влияния на активность катализатора Ni/Al2O3 различных солей Ni, используемых для его приготовления, показало, что в случае применения органических солей никеля (ацетилацетонат Ni и др.) формируется плотный углерод, который далее служит ядром для коксообразования. Это явление не возникает, если для приготовления катализатора используются неорганические соли Ni (нитраты, хлориды и др.). В работе [22] предложен новый метод получения катализатора Ni/Al2O3, включающий стадии осаждения углерода на поверхности и последующее удаление его по реакции с CO2. После такой многократной обработки уменьшается удельная поверхность Ni, но активность его растет, снижается углеотложение.
Согласно исследованию [22], высокая пористость Ni-катализаторов, нанесенных на Al2O3, SiO2, MgO, способствует повышению каталитической активности.
В работе [20] была изучена углекислотная конверсия метана при 650 °С и соотношении СН4:СО2 = 1:3 на катализаторе Ni/CaO-SiO2. Катализаторы готовили пропиткой солями Ni носителя SiO2, модифицированного СаО. При этом возрастает дисперсность металла.
Образующиеся угольные нити не дезактивируют катализатор.
Имеются данные о том, что дисперсность металла есть строгая функция кислотности носителя по Льюису [23]. Возможно, льюисовские центры являются центрами кристаллизации частиц металла.
По сообщениям [20, 21] регенерация в Н2 (700 °С, 12 ч) сильно увеличивает активность Ni-катализатора и уменьшает активность Ni-Co-катализатора. Изменения активности приписаны коксообразованию и структурным изменениям. Авторы [20, 21] делают вывод о том, что вклад паровой конверсии СО на этих катализаторах менее важен.
Никелевые катализаторы с добавками переходных металлов.
Исследовано влияние добавок La2O3, CeO2, а также оксидов MgO и CaO к катализатору Ni/Al2O3 на его активность и другие свойства в конверсии смеси CH4 + CO2 при 650─850 °С. Катализаторы, промотированные MgO и CaO, более чувствительны к условиям пропитки, чем катализаторы, промотированные оксидами редкоземельных элементов. Высокую активность проявляют катализаторы Ni/CeO2 и Ni/CeO2─Al2O3 [23]. Наиболее активен катализатор, содержащий 5% CeO2. Добавка способствует увеличению восстанавливаемости и диспергируемости никеля. Благодаря оксиду CeO2 после диссоциативной адсорбции CO2 атом углерода реагирует с кислородом и меньше образуется кокса.
Изучен катализатор Ni/MgO (Ni : Mg = 1:1) с добавками Cr2O3 и La2O3 [23]. Установлено, что введение Cr2O3 или La2O3 обеспечивает значительное повышение устойчивости катализатора к коксообразованию. Промотирование этими добавками увеличивает степень окисления Ni, что снижает склонность метана к глубокому дегидрированию (до углерода).
В Институте химической физики РАН проведено изучение широкого набора Ni/MgO-катализаторов с различными добавками [23]. Высокую активность показали Ni/MgO-катализаторы с добавками оксидов CeO2, CuO, Cr2O3, MnO2, которые в условиях катализа могут подвергаться восстановлению и окислению. Из них катализатор Ni/MgO-Cr2O3 оказался наиболее активным; при составе 6%Ni-1%Cr2O3-MgO он обеспечивает конверсию, близкую к равновесной, уже при ~ 700 °С (рис. 14). Как следует из зависимостей, представленных на рис. 14, даже небольшие количества Ni и Cr2O3 взаимно промотируют друг друга.
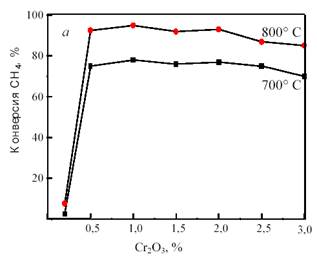
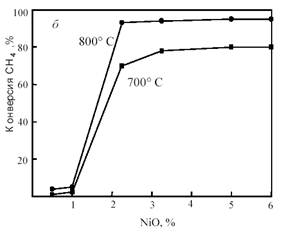
Рис. 14. Зависимость конверсии метана на катализаторе NiO-Cr2O3/MgO от содержания Cr2O3 при фиксированном содержании NiO (3%) (а) и от содержания NiO при фиксированном содержании Cr2O3 (2%) (б)
Была изучена углекислотная конверсия СН4 на Ni-катализаторах, нанесенных на α-Al2O3, γ-Al2O3, α-Al2O3•SiO2, ZrO2, MgO и модифицированных переходными металлами (Co, Cu, Fe), а также щелочными промоторами (Na, K). [23] Каталитическая активность Ni/α-Al2O3 очень близка к таковой для Ni/γ-Al2O3, но последний быстрее закоксовывается из-за его кислотных свойств. Для Ni-катализаторов на этих носителях получен следующий ряд их устойчивости к коксообразованию: α-Al2O3 > γ-Al2O3 > SiO2 > α-Al2O3•SiO2 > ZrO2, MgO. По силе влияние переходных металлов на катализатор Ni/α-Al2O3 соответствует ряду: Ni-Co, Ni > Ni-Cu >> Ni-Fe, а щелочных добавок ─ ряду: Ni > Ni-Na > Ni-K. Добавки металлов уменьшают восстанавливаемость никеля, но увеличивают его дисперсность. После 12 ч работы при 700 °С Ni-катализатор полностью дезактивировался, в то время как активность Ni-Co-катализатора сильно увеличилась при полном отсутствии коксообразования.
Оксид ZrO2 в катализаторе Ni/ZrO2-MgO стабилизирует состояние никеля на носителе MgO [23]. Температура восстановления NiO повышается с ростом количества MgO. Так, катализатор Ni/ZrO2 без MgO при 800 °С очень мало активен, добавление же всего 1% MgO приводит к 90%-ной конверсии метана, что близко к равновесию.
Высокоэффективными катализаторами сухой конверсии метана являются Ni-содержащие перовскиты LaNixFe(1─ x)О3, конверсия СН4 и СО2 составляет 97.5%, выход СО равен 97,1% при 800 °С [24]. В условиях катализа смешанная перовскитная структура разрушается, но при составе перовскита х < 0,5 катализаторы могут быть регенерированы путем прокаливания. При 0,3 < x < 0,8 образуются сплавы никеля с железом разного состава. Предполагается, что образование сплава предотвращает отравление катализатора углеродом благодаря торможению диффузии углерода сквозь частицу Ni.
Каталитическая активность и коксоустойчивость перовскитов состава La1─ xSrxNiO3 (x = 0, 0,1) и La2 ─ xSrxNiO3 (x = 0,1) при 600─900 °С и атмосферном давлении зависит от типа перовскита и степени замещения
Sr [24]. Так, LaNiO3 показывает высокую каталитическую активность, а La2NiO4 ─ совершенно неактивен. Среди стронций-замещенных катализаторов La0,9Sr0,1NiO3 и La1,8Sr0,2NiO4 имеют максимальную активность.
Начальная активность катализаторов этого типа растет со временем по мере их работы, достигая стационарного состояния. Рентгенофазовый анализ отработанных катализаторов показывает, что в условиях реакции катализаторы превращаются в смесь фаз La2O2CO3 и SrCO3 с высокодисперсным Ni. Такое превращение может быть вызвано удалением решеточного кислорода при замещении, ускоряемом в восстановительной атмосфере при CH4/CO2 = 1. Предположительно, высокая активность обусловлена двумя центрами: La2O3 служит для адсорбции CO2, Ni ─ для активации CH4.
Перовскитные катализаторы Ni/Ca0,8Sr0,2TiO3 и Ni/BaTiO3, приготовленные методом твердофазной кристаллизации, были испытаны в сухом риформинге метана. В условиях катализа образуется металлический Ni, который равномерно распределяется в решетке перовскитной матрицы. Отмечено, что внедрение Ni в решетку BaTiO3 происходит легче, чем в решетку Ca0,8Sr0,2TiO3. Высокая дисперсность никеля приводит к высокой активности и коксоустойчивости катализатора, причем снижение углеобразования частично обязано присутствию щелочноземельных металлов. Подвижный кислород в решетке перовскита также способствует удалению углерода.
Изучено влияние добавок Mo и W на каталитические свойства системы Ni/Al2O3 [24]. Никелевый катализатор, легированный малыми добавками Мо, дезактивируется, хотя при низких степенях легирования углерода на поверхности катализатора не наблюдалось. Напротив, катализатор, легированный W, не дезактивируется, углерода на поверхности значительно меньше, чем на непромотированном катализаторе. Ингибирование углеотложения объясняется образованием карбидов Мо и
W, активных в сухой конверсии метана. По-видимому, карбидные центры формируются вблизи никелевых центров в Ni/Al2O3, промотированном Mo/W. Эти центры обеспечивают диссоциацию СО2 и увеличивают на каталитической поверхности количество кислорода в атомарном состоянии, доступное для реакции с поверхностным углеродом.
4.5 Технология конверсии метана
Способ паровой конверсии в трубчатых печах применяется для получения синтез-газа, используемого для производства водорода, аммиака и метанола. Для синтеза метанола паровая конверсия обладает существенным недостатком ─ получают газ с избыточным содержанием водорода, а переработка таких газов приводит к увеличению затрат на сжатие. Кроме того, избыточный водород ─ балласт в процессе синтеза, и его приходится отводить с продувочными газами. Но, несмотря на это, процесс паровой конверсии все еще считается наиболее экономически эффективным вариантом для крупнотоннажных (750 тыс. т/год) установок производства метанола, имеющих одну технологическую линию и рассчитанных на использование в качестве сырья газа по низкой или умеренной цене.
В России наиболее широко распространен процесс паровой конверсии метана. Процесс идет в несколько стадий: подготовка сырья, конверсии, утилизации тепла, очистки газов от CO2. Сырье очищают по необходимости.
Исходный метан сжимают турбокомпрессором 1 до 2 ─ 3 МПа (см. рис. 15) и смешивают с необходимым количеством водяного пара и CO2. Смесь подогревают в теплообменнике 2 до 400 оС частично охлажденным конвертированным газом и подают в смеситель конвертора 6, куда поступает предварительно приготовленная смесь O2 с равным объемом водяного пара. Конвертор охлаждается кипящим в рубашке конденсатом; при этом генерируется пар с давлением 2 ─ 3 МПа, который отделяют в паросборнике 5. Тепло горячего конвертированного газа, выходящего из конвертора при 800 ─ 900 oC, используют в котле-утилизаторе для получения пара высокого давления, направляемого затем в линию пара соответствующего давления или используемого для привода турбокомпрессора. Тепло частично охлажденного газа утилизируют для предварительного подогревания смеси в теплообменнике 2 и в теплообменнике 3 для нагревания водного конденсата, питающего котел-утилизатор. Окончательное охлаждение осуществляют в скруббере 7 водой, циркулирующей через холодильник 8. При этом на выходе газ содержит смесь газов следующего состава:
CO ─ 15 – 45% (об.)
H2 ─ 40-75% (об.)
CO2 ─ 8-15% (об.)
CH4 ─ 0,5% (об.)
N2 и Ar ─ 0,5-1% (об.)
Очищают от CO2 через абсорбцию под давлением, хемосорбцию водным раствором моноэтаноламина или карбоната калия.
Наверх газ поступает в абсорбер 9, где поглощается CO2, а очищенный газ направляется к потребителю. Насыщенный абсорбент подогревается в теплообменнике 10 горячим регенерированным раствором и направляется в десорбер 11, с низа которого абсорбент направляется через т/о 10 вновь на поглощение CO2 в абсорбер 9. CO2 с верха 11 компримируют до соответствующего давления и возвращают на конверсию, смешивая перед т/о 2 с природным газом и водяным паром.
Расход на 1 м3 синтез-газа составляет:
Природный газ ─ 0,35 – 0,40 м3,
Технический O2 ─ 0,2 м3,
и в зависимости от применяемого
давления и добавки CO2 ≈ 0,2 - 0,8 кг водяного пара.
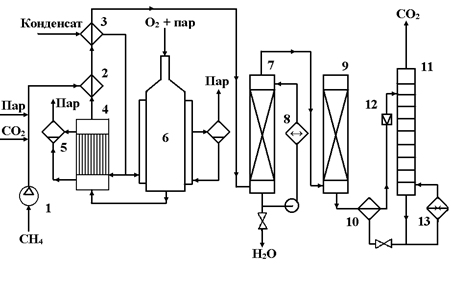
Рис. 15. Технологическая схема окислительной конверсии природного газа при высоком давлении
1 – турбокомпрессор; 2, 3, 10 – теплообменники; 4 – котел-утилизатор; 5 – паросборники; 6 – конвертор; 7 скруббер; 8 – холодильник; 9 – абсорбер; 11 – десорбер; 12 – дроссельный вентиль; 13 – кипятильник.
За рубежом развитие процессов паровой и углекислотной конверсии метана направлено несколько в другую сторону. На рис. 16 показаны принципиальные технологические схемы конверсии метана с паром для получения водорода и комбинированного парового/углекислотного риформинга для производства синтез-газа.
Установка риформинга обычно включает от 40 до 400 трубок (длина 6 ─ 12 м, диаметр 70 ─ 160 мм, толщина стенок 10 ─ 20 м), которые установлены вертикально в прямоугольной печи. Трубки заполняются катализатором, обычно формованным в виде небольших цилиндров или колец Рашига. Реактор обогревается горелками, которые могут размещаться внизу, сбоку или сверху печи. Топливо сжигается в радиационной секции печи. Отходящие дымовые газы (после обогрева реактора). Проходят через конвекционную секцию, где охлаждаются за счет отдачи тепла жидкостным и паровым потокам, включая пар, необходимый для реакции, исходный водяной поток и потоки для производства пара.
Дальнейший технологический маршрут синтез-газа зависит от выбранного процесса его вторичной переработки (получение H2, CO, оксосинтез, синтез аммиака и т.д.). Для получения водорода газ направляют в реактор конверсии водяного газа и поглотительный реактор переменного давления (см. рис.16а). Если требуется получить CO, то используется технологическая схема, включающая секцию удаления CO2 и установку низкотемпературного разделения (т.н. «холодный ящик»). Выделяемый диоксид углерода повторно используется в процесс риформинга. Если в этом случае желательно также получить H2 в качестве продукта, то установку снабжают блоком PSA (Pressure-Swing-Adsorption) (см. рис. 16).
В таблице 6 представлены некоторые характеристик процессов конверсии метана.
Таблица 6
Сравнительные технико-экономические показатели процессов получения синтез-газа.
| Показатели |
Паровая конверсия углеводородного газа |
Двухступенчатая конверсия в системе конвертеров "Тандем" |
Паро-углекислотная конверсия природного газа |
|
Сырье, требования к сырью |
Природный газ, легкая часть попутного нефтяного газа |
Природный газ, легкая часть попутного нефтяного газа Кислород - 138 н.м3/1000 н. м3 |
Природный газ, легкая часть попутного нефтяного газа CO2 из отделений синтеза |
| Получаемые продукты |
Синтез-газ, % (об.) CO2 - 7,59 CO - 14,28 H2 - 73,02 CH4 - 4,67 N2 - 0,51 Калорийность 2688 ккал/н.м3 Пар высокого давления - 1,31 т/1000 н.м3 синтез-газа |
Синтез-газ, % (об.) CO2 - 7,49 CO - 22,03 H2 - 67,42 CH4 - 2,17 N2 - 0,89 Калорийность 2578 ккал/н.м3 Пар высокого давления - 0,5 т/1000 н.м3 синтез-газа |
Синтез-газ, % (об.) CO2 - 0,04 CO - 31,27 H2 - 36 CH4 - 2,28 |
|
Расходные показатели (на 1000 н.м3 синтез-газа) |
Природный газ: на технологию - 261 н.м3 на топливо - 47 н.м3 всего - 308 н.м3 Катализаторы - 0,209 кг |
Природный газ: на технологию - 312 н.м3 на топливо - 54 н.м3 всего - 366 н.м3 Катализаторы - 0,195 кг |
Природный газ: на технологию - 261 н.м3 на топливо - 82 н.м3 всего - 343 н.м3 Катализаторы - 0,2 кг |
|
Влияние на окружающую среду (на 1000 н.м3 синтез-газа) |
Дымовые газы огневого подогревателя - 113 н.м3 NOx + SO2 = 0,006 кг Дымовые газа после трубчатой печи - 2580 н.м3 NOx + SO2 = 0,085 кг Отработанные катализаторы |
Дымовые газы вспомогательного котла – 36 н.м3 Отработанные катализаторы |
Дымовые газы - 2290 н.м3 Отработанные катализаторы |
Таблица 6 (окончание)
Технико-экономические показатели.
| Показатели |
Паровая конверсия углеводородного газа |
Двухступенчатая конверсия в системе конвертеров "Тандем" |
Паро-углекислотная конверсия природного газа |
|
Выход CO на исходный углерод, % |
53 - 55 | 69 - 70 | 93 |
|
Соотношение H2:CO,моль/моль |
5:1 | (3,0 - 3,2):1 | (2,1 - 2,3):1 |
|
Удельные кап. вложения, долл. /1000 н.м3 газа в год |
73,4 | 78,2 | 99,9 |
| Расход природного газа на 1000 н.м3 синтез-газа | 308 | 366 | 343 |
|
Давление получаемого синтез-газа, МПа |
1,2 - 1,6 | 2,8 - 3,0 | 1,2 - 1,5 |
5. Синтез Фишера-Тропша
Метод Фишера – Тропша по превращению метана в более тяжелые углеводороды был разработан в 1923 г. и реализован в промышленности Германии в 1940-х годах.
Почти все авиационное топливо в этой стране во время второй мировой войны производилось с помощью синтеза Фишера – Тропша из каменного угля. Впоследствии от этого способа изготовления моторных топлив отказались, так как топливо, получаемое при переработке нефти, до последнего времени было экономически более выгодным.
При получении жидкого топлива на основе синтеза Фишера - Тропша разнообразные соединения углерода (природный газ, каменный и бурый уголь, тяжелые фракции нефти, отходы деревообработки) конвертируют в синтез-газ (смесь СО и Н2), а затем он превращается в синтетическую «сырую нефть» - синтнефть. Это – смесь углеводородов, которая при последующей переработке разделяется на различные виды практически экологически чистого топлива, свободного от примесей соединений серы и азота. Достаточно добавить 10% искусственного топлива в обычное дизельное, чтобы продукты сгорания дизтоплива стали соответствовать экологическим нормам. [28]
Еще более эффективной представляется конверсия газа в дорогостоящие продукты тонкого органического синтеза.
Конверсию газа в моторное топливо можно в целом представить как превращение метана в более тяжелые углеводороды:
2nСН4 + 1/2nО2 = СnН2n + nН2О
Из материального баланса брутто-реакции следует, что массовый выход конечного продукта не может превышать 89%.
Реакция напрямую неосуществима. Конверсия газа в жидкое топливо (КГЖ) проходит через ряд технологических стадий (рис.17). При этом в зависимости от того, какой конечный продукт необходимо получить, выбирается тот или иной вариант процесса.
Синтез Фишера-Тропша может рассматриваться как реакция восстановительной олигомеризации монооксида углерода, при которой образуются углерод-углеродные связи, и в общем виде она представляет собой сложную комбинацию ряда гетерогенных реакций, которую можно представить суммарными уравнениями:
nCO + 2nH2 → (CH2)n + nH2O,
2nCO + nH2 → (CH2)n + nCO2 .
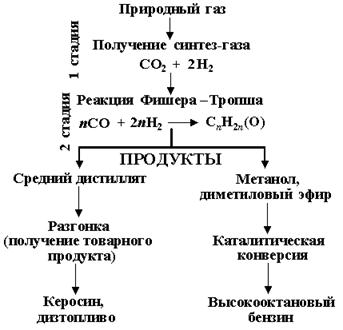
Рис. 17. Принципиальная схема конверсии синтез-газа в жидкие продукты (топливо).
Продуктами реакции являются алканы, алкены и кислородсодержащие соединения, то есть образуется сложная смесь продуктов, характерная для реакции полимеризации. Первичными продуктами синтеза Фишера-Тропша являются a- и b-олефины, которые превращаются в алканы в результате последующего гидрирования. Природа применяемого катализатора, температура, соотношение СО и Н2 существенно сказываются на распределении продуктов. Так, при использовании железных катализаторов велика доля олефинов, тогда как в случае кобальтовых катализаторов, обладающих гидрирующей активностью, преимущественно образуются насыщенные углеводороды. [2]
В настоящее время в качестве катализаторов синтеза Фишера-Тропша в зависимости от поставленных задач (повышение выхода бензиновой фракции, увеличение выхода низших олефинов и др.) используются как высокодисперсные железные катализаторы, нанесенные на оксиды алюминия, кремния и магния, так и биметаллические катализаторы: железо-марганцевые, железо-молибденовые и др. [34]
За 70 лет с момента открытия синтеза не утихают споры по поводу механизма реакции. В настоящее время рассматриваются три различных механизма. Первый механизм, называемый карбидным, впервые предложенный Фишером и Тропшем и в дальнейшем нашедший поддержку у других исследователей, предполагает образование С-С-связей в результате олигомеризации метиленовых фрагментов на поверхности катализатора. На первой стадии происходит адсорбция СО и образуется поверхностный карбид, а кислород превращается в воду или СО2:
На второй стадии поверхностный карбид гидрируется с образованием фрагментов СНx (х = 1-3):

Удлинение цепи происходит в результате реакции поверхностных метила и метилена и далее путем внедрения метиленовых групп идет рост цепи:
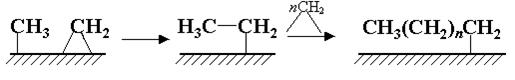
Стадия обрыва цепи происходит в результате десорбции алкена с поверхности катализатора:
Второй механизм, названный гидроксикарбеновым, предполагает также гидрирование координированного на металле СО с образованием поверхностных гидроксикарбеновых фрагментов, в результате конденсации которых и происходит образование С-С-связей:
Третий механизм, который можно назвать механизмом внедрения, предполагает образование С-С-связей в результате внедрения СО по связи металл-углерод (о способности СО к внедрению по связи металл-алкил говорилось выше):
Накоплен достаточно богатый экспериментальный материал, свидетельствующий в пользу того или иного варианта механизма, однако приходится констатировать, что к настоящему моменту невозможно сделать однозначный выбор между ними. Можно предположить, что в связи с большой важностью синтеза Фишера-Тропша исследования в этом направлении будут интенсивно продолжаться и мы станем свидетелями новых воззрений на механизмы протекающих реакций [3].
5.1 Выбор катализаторов
Состав конечных продуктов зависит от катализатора, температуры и соотношения СО и Н2.
На металлоокисном катализаторе получают метанол с примесью этанола и диметилового эфира. Это основной процесс получения метанола в мире, обычная мощность метанольных заводов составляет около 0,5 млн. т в год (Новомосковское ПО «АЗОТ»; кобальтовый катализатор).
Для производства моторных топлив метанол перерабатывается в диметиловый эфир и далее в смесь разветвленных предельных углеводородов (процесс Mobil GTG в Мауи, Новая Зеландия; кобальтовый катализатор)[19].
На кобальтово-цинковых катализаторах, обладающих гидрирующей активностью, получают смесь линейных алканов (процесс AGC-211 в Бинтулу, Малайзия) [20].
На железном катализаторе получают смесь линейных и разветвленных алканов и алкенов (перспективный процесс Рентех).
На кобальтовых или родиевых катализаторах при давлении выше 10 МПа и температуре в диапазоне 140 - 180 °С алкены взаимодействуют с синтез-газом и превращаются в альдегиды - важнейшие полупродукты в производстве спиртов, карбоновых кислот, аминов, многоатомных спиртов и др. Мировое производство альдегидов по такой технологии (оксосинтез) достигает 7 млн т в год [14].
Одно из важных современных направлений научного поиска в области синтеза Фишера - Тропша состоит в получении кислородсодержащих продуктов. Введение таких соединений в количестве 1 % в дизельное топливо снижает содержание сажи в продуктах сгорания на 4 – 10%.
6. Альтернативный способ получения синтез-газа
Современные проблемы энергетики могут быть решены только при рациональном использовании всех существующих на Земле и околоземном пространстве источников топлива и энергии [29]. Среди них биомасса, как постоянно возобновляемый источник топлива, занимает существенное место [32].
Биомасса - термин, объединяющий все органические вещества растительного и животного происхождения. Биомасса делится на первичную (растения, животные, микроорганизмы и т.д.) и вторичную - отходы при переработке первичной биомассы и продукты жизнедеятельности человека и животных. В свою очередь отходы также делятся на первичные - отходы при переработке первичной биомассы (солома, ботва, опилки, щепа, спиртовая барда и т.д.) и вторичные - продукты физиологического обмена животных и человека.
Ежегодное количество органических отходов по разным отраслям народного хозяйства России составляет более 390 млн. т. Сельскохозяйственное производство дает 250 млн. т, из них 150 млн. т приходится на животноводство и птицеводство, 100 млн. т -на растениеводство. Лесо- и деревопереработка дают 700 млн. т, твердые бытовые отходы городов - 60 млн. т, коммунальных стоков - 10 млн. т (все приведенные значения даются на абсолютно сухое вещество.
Энергия, запасенная в первичной и вторичной биомассе может конвертироваться в технически удобные виды топлива или энергии несколькими путями.
1. Получение растительных углеводородов (растительные масла, высокомолекулярные жирные кислоты и их эфиры, предельные и непредельные углеводороды и т.д.). Например, для южных регионов России это может быть рапсовое масло, добавляемое к дизельному топливу.
2. Термохимическая конверсия биомассы (твердой, до 60%) в топливо: прямое сжигание, пиролиз, газификация, сжижение, фест-пиролиз.
3. Биотехнологическая конверсия биомассы (при влажности от 75 % и выше) в топливо: низкоатомные спирты, жирные кислоты, биогаз.
В зависимости от влажности биомасса перерабатывается термохимическими или биологическими способами. Биомасса с низкой влажностью (сельскохозяйственные и городские твердые отходы) перерабатываются термохимическими процессами: прямым сжиганием, газификацией пиролизом, ожижением, гидролизом. В результате получают водяной пар, электроэнергию, топливный газ, водород (метанол), жидкое топливо, газ, древесный уголь, глюкоза.
Биомасса с высокой влажностью (сточные воды, бытовые отходы, продукты гидролиза органических остатков) перерабатываются биологическими процессами: анаэробная переработка, этанольная ферментация, ацетонобутанольная ферментация. В результате этих процессов получают биогаз (СН4, СО2), органические кислоты, этанол, ацетон, бутанол. Различие физико-химических свойств биомассы обусловливает выбор термохимического или биологического процесса ее переработки.
6.1 Термохимическая конверсия биомассы
Прямое сжигание является одним из самых широко применяемых методов переработки биомассы (древесины и древесных отходов, соломы, городских твердых отходов и др.).
Топливо, вырабатываемое из городских твердых отходов, используют в сочетании с углем на небольших электростанциях.
Наиболее перспективными и все более широко применяемыми процессами превращения биомассы в различные виды энергии являются термохимическая газификация, этанольная ферментация и анаэробная переработка. Из термохимических процессов переработки биомассы наибольшее внимание в настоящее время привлекают такие, как газификация, пиролиз и сжижение, в результате которых получают жидкие и газообразные топлива, имеющие значительно большую энергоемкость, чем биомасса. Все эти процессы протекают при высокой температуре, а иногда и при высоком давлении.
Газификация древесины и другого лигноцеллюлозного сырья в течение многих лет является одним из основных методов производства низкокалорийного топливного газа. Топливный газ может быть непосредственно использован в котельных, обжигательных печах и разного вида топках, а после охлаждения, очистки и осушки - в качестве топлива в двигателях внутреннего сгорания. Состав получаемых при газификации газов зависит от природы применяемого сырья, типа окислителя, температуры процесса и давления. Наибольшую ценность представляет среднекалорийный газ, особенно синтез-газ (в основном состоящий из СО и Н2). При газификации древесины получают синтез-газ, который по составу идентичен синтез-газу, вырабатываемому газификацией угля, паровой конверсией природного газа и др.
Пиролиз биомассы осуществляется при ее нагревании в отсутствии кислорода с образованием жидкого топлива, газов и древесного угля. Выход продуктов пиролиза зависит от условий проведения процесса и типа сырья. В свою очередь, условия процесса определяются природой сырья, заданными продуктами производства.
Широкую известность получил процесс превращения биомассы в жидкое топливо пиролизом со ступенчатым испарением, где в качестве сырья используются твердые городские отходы, древесная кора и др.
Основными технологическими узлами установки являются отделения для предварительной обработки древесины, производства синтез-газа, реакторное и секция для разделения продуктов ожижения. В отделении для предварительной обработки древесины биомасса (в виде древесной щепы) высушивается, измельчается и смешивается с рециркулирующей частью производимого жидкого топлива. Полученная суспензия нагревается до 200 оС и под давлением 23 МПа подается в реактор, где в присутствии раствора углекислого натрия в качестве катализатора и смеси газов оксида углерода и водорода (поступающего из отделения производства синтез-газа после очистки последнего от СО2 и Н2О) при температуре 340 оС и давлении 23МПа происходит ожижение биомассы.
Неочищенная жидкая фракция содержит, кроме образовавшегося жидкого топлива, непрореагировавшую древесину, катализатор и нерастворимые твердые вещества, для очистки от которых она направляется в сепараторное отделение. Извлеченные из неочищенного жидкого топлива твердые вещества и водорастворимый катализатор возвращают в систему. Общий тепловой КПД промышленной установки (с учетом всех потерь) составляет 50-60 %.
Один из методов переработки целлюлозной биомассы (например, соломы) - гидролиз минеральными кислотами с образованием глюкозы и ксилозы, которые в дальнейшем могут быть подвергнуты ферментации в целях производства различных органических химикатов, включая этанол, кислоты, бутанол и ацетон. С точки зрения получения заменителей жидкого и газообразного ископаемого топлива наибольший интерес представляет технология переработки биомассы с образованием в качестве конечных продуктов этанола, метанола, синтетического природного газа и биогаза.
Основными преимуществами превращения биомассы методом термохимической газификации являются высокие эффективность и скорость превращения. К недостаткам процесса относится возможность переработки сырья только с низким содержанием влаги, а также высокие температура и давление, сложные техническое оформление и управление процессом.
6.2 Биотехнологическая конверсия биомассы
При биотехнологической конверсии, как правило, используется биомасса и прежде всего разнообразные органические отходы с влажностью не менее 75%, хотя в России разработаны научные основы биоконверсии биомассы с влажностью менее 75% - твердофазная метангенерация осадков сточных вод и твердых бытовых отходов [32].
Биологическая конверсия биомассы в топливо и энергию развивается по двум основным направлениям:
· ферментация с получением этанола, низших жирных кислот, углеводородов, липидов - это направление давно и успешно используется на практике;
· получение биогаза.
В настоящее время получение биогаза связано прежде всего с переработкой и утилизацией отходов животноводства, птицеводства, растениеводства, пищевой, спиртовой промышленности, коммунально-бытовых стоков и осадков.
В России имеются технологии по переработке отходов птицеводства. Одна из них, разработанная Т. Я. Андрюхиным, введена в эксплуатацию в 1987-1988 гг. на Октябрьской птицефабрике Глебовского ППО Истринского района Московской области. Станция перерабатывает ежесуточно 10 т куриного помета механической уборки и производит до 1000 м3 биогаза и до 30 т экологически чистых органических удобрений [33].
Из биологических методов превращения биомассы наибольшее распространение получают анаэробная переработка и этанольная ферментация. В процессе анаэробной переработки или перегнивання (метановая ферментация) органические вещества разлагаются до СО2 и CH4. Возможность получения высококалорийного, топливного газа (СН4) путем биохимической переработки биомассы, частности экскрементов крупного рогатого скота, реализована сравнительно недавно. Процесс анаэробной переработки органических отходов происходит в отсутствии кислорода с участием различных групп бактерий.
Одним из способов переработки биомассы является ацетонобутанольная ферментация, в результате которой под действием микроорганизма образуется уксусная и масляная кислота, этанол, бутанол, ацетон, изопропанол, а также диоксид углерода и водород.
7. Продукты, получаемые на основе синтез-газа
Альтернативные технологии получения качественных моторных топлив включают стадии газификации твердого сырья в смесь CO и H2 и последующего синтеза углеводородных смесей, используемых в качестве бензина, дизельного топлива или компонентов моторных топлив по схеме (рис. 18).
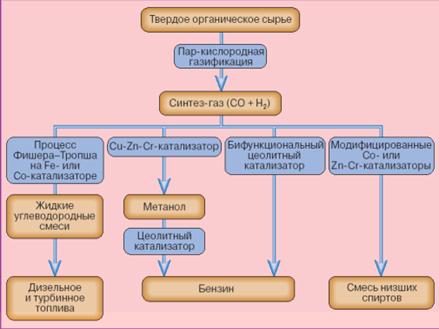
Рис. 18. Схема получения продуктов из твердого органического сырья через синтез-газ.
Жидкие продукты процесса Фишера-Тропша, образующиеся из синтез-газа на промотированных железных или кобальтовых катализаторах, содержат преимущественно неразветвленные парафиновые углеводороды. Фракции этих жидких продуктов могут использоваться в качестве дизельных и турбинных топлив с минимальной переработкой.
Осуществление процесса Фишера – Тропша в жидкой фазе с использованием суспензии катализатора дает возможность перерабатывать синтез-газ с высоким содержанием CO в качественные жидкие топлива. Применение синтез-газа с высоким отношением CO\H2 позволяет исключить стадию конверсии CO водяным паром, которая обычно используется для получения дополнительного количества Н2, и повысить термическую эффективность процесса.
В таблице 7 представлены данные о продуктах, получаемых на основе синтез-газа.
Таблица 7.
Характеристика производства различных продуктов, получаемых на основе синтез-газа.
| Процесс | Продукт | Состав исходного газа |
Расход 1 т конечного продукта |
Затраты тв. топливана 1 т конечного продукта |
| Синтез аммиака | аммиак |
75% (об) H2, 25% (об) N2 |
2050 м3 H2+ 685 м3 N2 |
1,40 |
| Синтез метанола | метанол |
67% (об) Н2, 33% (об) CO |
1650 м3 H2+ 825 м3 CO |
1,50 |
| Оксосинтез | альдегиды, спирты |
50% (об) H2, 50%(об) CO |
600 м3 H2 + 600 м3 CO |
0,88 |
|
Синтез у/в по Фишеру-Тропшу |
жидкие углеводороды |
33% (об) Н2, 67% (об) или 67% (об) Н2, 33% (об) CO |
2000 м3 H2+ 4000 м3 CO или 4000 м3 H2 + 2000 м3 CO |
3,85 |
|
Прямое восстановление железа |
железная губка (92%Fe) |
33% (об) Н2, 67% (об) |
225 м3 H2 + 400 м3 CO |
0,45 |
|
Гидрокрекинг вакуумного дистиллята нефти |
бензин |
100% (об) H2 |
500 м3 H2 |
0,02 |
|
Гидрирование каменного угля |
жидкие углеводороды |
100% (об) H2 |
2070 м3 H2 |
0,27 |
|
Гидрирование бурого угля |
жидкие углеводороды |
100% (об) H2 |
1620 м3 H2 |
0,16 |
8. Заключение
В мире проводятся интенсивные исследования по усовершенствованию и замене традиционных методов получения синтез-газов на более современные, с целью снижения капиталовложений и эксплуатационных затрат на этой стадии.
В настоящее время удельные капитальные затраты производства моторных топлив из природного газа через стадию получения синтез-газа и синтез Фишера-Тропша почти в 2 раза выше, чем у процессов переработки нефти.
Перспективными являются работы по синтезу метанола и водородного топлива через процесс получения синтез-газа.
Из изученных материалов можно сделать следующие заключения:
· наиболее выгодным процессом крупнотоннажного получения синтез-газа остается паровая конверсия метана;
· самым дорогим процессам по капиталовложениям является паро-углекислотная конверсия метана, которая в то же время дает наибольший выход монооксида углерода;
· при сравнении 3-х методов газификации видно, что Koppers-Totzek практически по всем показателям превосходит методы Lurgi и Winkler;
· для экологической безопасности необходимо улучшать системы улавливания вредных примесей, а также подвергать более полной утилизации отработанные катализаторы.
9. Литература
1. Катализ в С1 – химии. / Под ред. Л. Кайма. Л.: Химия, 1987. 296 с.
2. Караханов Э. А., Что такое нефтехимия // Соросовский Образовательный журнал. 1996. № 2. С. 65─73.
3. Харитонов Ю. Я. Комплексные соединения // Соросовский Образовательный журнал. 1996. № 1. С. 48─56.
4. Химические вещества из угля. Пер. с нем./ Под ред. Э. Фальбе – М: Химия, 1980. ─ 616 с.
5. Бекаев Л. С., Марченко О. В., Пинегин С. П. и др. Мировая энергетика и переход к устойчивому развитию – Новосибирск: Наука, 2000. ─ 300с.
6. Шиллинг Г.-Д., Бонн Б.. Краус У. Газификация угля / Пер. с нем. и ред. С. Р. Исламова – МЖ Недра, 1986 – 175 с.
7. Патент 2052492 РФ. Способ получения синтез-газа и газификатор вертикального типа / С. Р. Исламов, С. Г. Степанов, А. Б. Морозов, О. С. Пивоваров, В. А. Збруев. – Опубл. 20. 01.1996 г. в БИ № 2. – 4 с.
8. Патент 2144844 РФ. Катализатор (его варианты) и процесс получения синтез-газа / Павлова С.Н., Сапутина Н.Ф., Садыков В.А., Бунина Р.В., Исупов В.П. ─ Опубл. 27.01.2000 в БИ № 9. – 13 с. Патентообладатель: Институт катализа им. Г. К. Борескова СО РАН
9. Dybkjaer J., Hansen J.B. Proc. IV Int. Natural Gas Conversion Symp. Kruger National Park, South Africa, 1995. Amsterdam: Elsevier. 1997, p. 99 ─116.
10. Fleisch T.N., Basu A., Gradassi M.J., Masin J.C. Ibid., p. 117.
11. Бодров И. М., Апельбаум Л. О. Кинетика и катализ, 1967, т. 8, № 4, с. 379─384.
12. Bradford M.C.J., Vannice M.A. Catal. Today, 1999, v. 50, № 1, p. 87─96.
13. Nakamura J., Aikawa K., Sato K., Uchijima T. Catal. Lett., 1994, v. 29, p. 261.
14. Крылов О.В., Мамедов А.Х. Успехи химии, 1995, т. 64, № 9, с. 935─959.
15. Erdohelyi A., Fodor K., Solymosi F.. Proc. IV Int. National Gas Conversion Symp. Kruger National Park, South Africa, 1995. Amsterdam: Elsevier, 1997, p. 525─530.
16. Kroll V.C.H., Tjatjopoulos G.J., Mirodatos C. Proc. V Int. Natura Gas Conversion Symp. Giardini-Naxos, Sicily, 1998. Amsterdam Elsevier, 1998, p. 753─758.
17. Schuurman Y., Marquez-Alvarez C., Kroll V.C.H., Mirodatos C. Catal. Today, 1998, v. 46, № 2.3, p. 185─192.
18. Gronchi P., Centola P., Kaddouri A., Del Rosso R. Proc. V Int. Natural Gas Conversion Symp. Giardini Naxos, Sicily, 1998. Amsterdam: Elsevier, 1998, p. 735─740.
19. Бобров Н.Н., Боброва И.И., Собянин В.А. Кинетика и катализ, 1993, т. 34, № 4, с 257─258.
20. Bobrova I.I., Bobrov N.N., Davydov A.A. Catal. Today, 1995, v. 24, № 3, p. 429─439.
21. Tomishige K., Chen Y., Yamazaki O. e.a. Proc. V Int. Natural Gas Conversion Symp. Giardini-Naxos, Sicily, 1998. Amsterdam: Elsevier, 1998, p. 861─866.
22. Томишиге К., Химено И., Ямазаки О. и др. Кинетика и катализ 1999, т. 40, № 3, с. 432─439.
23. Исаев О.В., Корчак В.Н., Крылов О.В. и др. Кинетика и катализ 2000, т. 45, № 11, с. 178─201.
24. O.Connor A.M., Ross J.R.H. Abstr. 5-th European Workshop on Methane Activation. Linerik, Ireland, 1997.
25. Содесава Т. Кинетика и катализ, 1998, т. 40, № 3, с. 452─453.
26. Krylov O.V., Mamedov A.Kh., Mirzabekova S.R. Catal. Today, 2000 г. v. 32, № 4, p. 173─189.
27. Бодров И.М., Апельбаум Л.О., Темкин М.И. Кинетика и катализ, 1998, т. 5, № 4, с. 696─703.
28. Н. И. Курбатов, А. К. Зайце ─ Конверсия природного газа в жидкое топливо // журнал «Потенциал», 1996. № 11. С. 44─52.
29. Панцхава Е.С., Кошкин Н.Л. Использование энергии биомассы в России: Проблемы и перспективы // Тезисы германо-российской конференции "Возобновляемые источники энергии и их роль в энергетической политике России и Германии". 1994. 24-26 октября, Фрайбург.
30. Кузнецов Б. Н. Новые подходы в химической переработке углей // Соросовский Образовательный Журнал. 1996. № 6. С. 50─58.
31. Шелдон Р. А. Химические продукты на основе синтез-газа: Пер. с англ. М.: Химия, 1987.
32. Березин И.В., Панцхава Е.С. Техническая биоэнергетика // Биотехнология. 1986. Т. 2. № 2. С. 1-12; № 3. С. 8-15. Панцхава Е.С., Давиденко Е.В. Метангенерация твердых органических отходов городов // Биотехнология. 1990. Т. 6. №4. С. 49-53.
33. Ре-циркуляционное анаэробное сбраживание отходов сельского хозяйства с выработкой биогаза / Т.Я. Андрюхин, Н.К. Свириденко, Ю.В. Савельев и др. // Биотехнология. 1989. Т. 5. №2. С. 219-225.
34. Караханов Э. А. Синтез-газ как альтернатива нефти. 1. Процесс Фишера-Тропша и оксо-синтез // Соросовский Образовательный Журнал. 1997. № 6. С.69.
| Шпаргалка по химии | |
|
01. Энтальпия и т.д 1. Н2 в природе. Изотопы Н2: протий, дейтерий, тритий. 2. Хим. св-ва Н2 3. Гидриды Ме и Нем, их св-во и получ. 4. Получение и прим ... 4. Получ Н2 В пром Н2 получают в основном из природных и попутных газов, продуктов газификации топлива (водяного и паровоздушного газов) и коксового газа. В основе производства водорода лежат каталитические реакции взаимодействия с водяным паром (конверсии) соотвнтственно углеводородов (главным образом метана) и Оха (II) углерода ... |
Раздел: Рефераты по химии Тип: реферат |
| Разработка предложений по очистке природного газа и переработки кислых ... | |
|
содержание Введение 1. Общие сведения о предприятии 1.1 Природно-климатическая характеристика района расположения предприятия 1.2 Характеристика ... Двуокись углерода СО2 - бесцветный, тяжелый, малореакционноспособный газ. СО2 + Н2S ° CO + Н2О + СОS (4.6) |
Раздел: Рефераты по экологии Тип: дипломная работа |
| ... в курсе школьного предмета химии на предмете углерода и его соединений | |
|
Приложение 1 Конкретные примеры о методах реализации межпредметных связей. 1. Вопросы межпредметного содержания: а) Вспомните (из курса географии ... При открытых разработках угля, подземной его газификации, получение угольного концентрата, сжигание угля на ТЭС в атмосферу выбрасывается, помимо частиц углерода, СО, СО2 ... Миллиарды тонн углекислого газа ежечасно поступают в атмосферу в результате сжигания угля и нефти, природного газа и дров, миллионы тонн метана поднимаются в атмосферу от ... |
Раздел: Рефераты по химии Тип: реферат |
| Метод дегазации угольных шахт с помощью сепаратора СЦВ-7 | |
|
СОДЕРЖАНИЕ СОДЕРЖАНИЕ ВВЕДЕНИЕ ГЛАВА 1. РОЛЬ МЕТАНА В УГОЛЬНОЙ ПРОМЫШЛЕННОСТИ ГЛАВА 2 . АНАЛИЗ ОБЪЕКТА ИССЛЕДОВАНИЯ 2.1 Анализ угольной промышленности ... По оценкам ведущих специалистов, метан имеет колоссальный потенциал парникового газа, превышающий в 21 раз двуокись углерода - основного соединения в индустриальных выбросах. Угольный метан в пересчете на условное топливо занимает 3 - 4 место в мире после угля, нефти и природного газа. |
Раздел: Рефераты по геологии Тип: дипломная работа |
| Программа для поступающих в вузы (ответы) | |
|
Программа по химии для абитуриентов Предмет химии. Явления химические и физические. Атомно-молекулярное учение. Атомы. Молекулы. Молекулярное и ... Из метана получают ценные химические продукты: метанол, синтез-газ, формальдегид, ацетилен, различные хлорпроизводные. Кроме метана природный газ содержит еще и другие углеводороды, а также азот, СО2, и часто - сероводород. |
Раздел: Рефераты по химии Тип: реферат |