Контрольная работа: Применение потенциометрического и кулонометрического методов анализа в фармации и аналитической химии
1. Смешаны 400 мл 0,0405 моль/л раствора бромата калия и 250 мл раствора бромата калия с молярной концентрацией эквивалента KВrO3 0,222 моль/л. Объем смеси разбавлен водой до 1000 мл. рассчитать молярную концентрацию эквивалента полученного раствора.
![]() 400мл – 0,0405 моль/л Ход решения:
400мл – 0,0405 моль/л Ход решения:
250мл – 0,222 моль/л Первым делом определим, сколько молей вещест-
![]() ва содержится в 400 мл
раствора и в 250 мл.
ва содержится в 400 мл
раствора и в 250 мл.
0,0405 моль – в 1000 мл
х - в 400 мл, следовательно
х=0,0405*400/1000=0,0162 моль
0,222 моль – в 1000 мл
у - в 250 мл
у=0,222*250/1000=0,0555 моль
Значит, в растворе будет содержатся 0,0162+0,0555=0,0717 моль вещества.
Поскольку раствор разбавляют водой до 1000мл (1л), получим, что молярная концентрация полученного раствора составит 0,0717 моль/л.
Теперь определим молярную концентрацию эквивалента полученного раствора.
Сн=nэ/Vp-pa, в свою очередь
nэ=m/Mэ, где Mэ – молярная масса эквивалента.
Молярная масса эквивалента соли определяется отношением молярной массы соли к произведению чиста атомов металла на его валентность. Поскольку К одновалентен и в данной соли содержится всего один его атом, следовательно Mэ=М, а значит молярная концентрация и молярная концентрация полученного раствора будут равны. Т.е. молярная концентрация эквивалента полученного раствора равна 0,0717 моль/л.
Ответ: 0,0717 моль/л.
2. Применение потенциометрического и кулонометрического методов анализа в фармации и аналитической химии.
Потенциометрический метод – это метод качественного и количественного анализа, основанный на измерении потенциалов, возникающих между испытуемым раствором и погруженным в него электродом. Данный метод рекомендуется для установления доброкачественности и количественного анализа некоторых фармакопейных препаратов. Использую потенциометрическое титрование, можно более объективно устанавливать точку эквивалентности, поэтому метод находит широкое практическое применение. Одним из направления потенциометрического метода является хронопотенциометрия. Сущность этого метода заключается в том, что потенциал одного их электродов записывают как функцию времени. Помимо аналитических целей метод может быть использован для изучения кинетики химических процессов. Потенциометрический метод также может быть использован при исследовании процессов разрушения лекарственных веществ при хранении.
Кулонометрический метод весьма перспективен для анализа лекарственных веществ: некоторых местноанестезирующих средств, сульфаниламидов, алкалоидов. Кулонометрический метод основан на законе Фарадея, устанавливающем связь между количеством вещества, выделившегося на электродах, и затраченным на этот процесс количеством электричества.
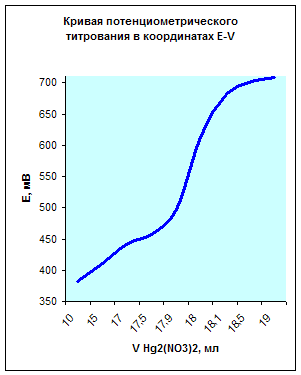
3. Построить кривые потенциометрического титрования в координатах E-V и ∆E/∆V-V и рассчитать концентрацию хлорида кальция в растворе (г/л), если при титровании 20,0 мл анализируемого раствора 0,0500 н. раствором Hg2(NO3)2 получили следующие данные:
|
V Hg2(NO3)2, мл |
10,0 | 15,0 | 17,0 | 17,5 | 17,9 | 18,0 | 18,1 | 18,5 | 19,0 |
| Е, мВ | 382 | 411 | 442 | 457 | 498 | 613 | 679 | 700 | 709 |
По данным таблицы получим следующую кривую потенциометрического титрования в координатах E-V:
|
V Hg2(NO3)2, мл |
10,0 | 15,0 | 17,0 | 17,5 | 17,9 | 18,0 | 18,1 | 18,5 | 19,0 |
| ∆E/∆V | 5,8 | 15,5 | 30 | 102,5 | 1150 | 660 | 52,5 | 18 |
Исходя из данных этой таблицы получим следующий график:
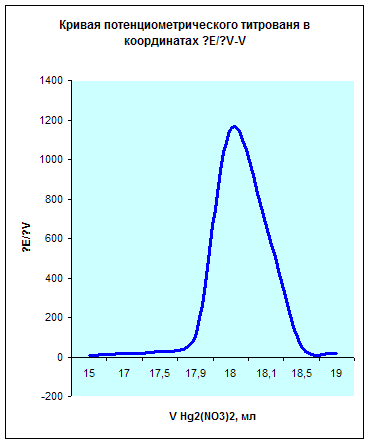
Из данных графиков видно, что точка эквивалентности соответствует 18мл Hg2(NO3)2. Отсюда можно рассчитать концентрацию хлорид кальция в растворе.
Уравнение реакции имеет следующий вид:
CaCl2+ Hg2(NO3)2® Hg2Cl2+Ca(NO3)2
m(Hg2(NO3)2)=Cн(Hg2(NO3)2)*Mэ(Hg2(NO3)2)*V(Hg2(NO3)2)
Нормальность данного раствора дана в условии задачи, объем определили по графику, осталось рассчитать Mэ.
Молярная масса эквивалента соли равна отношению мольной массы соли к произведению числа атомов металла на его валентность. Т.е., получим:
Mэ(Hg2(NO3)2)=![]() =131,5г/моль. Следовательно,
=131,5г/моль. Следовательно,
m(Hg2(NO3)2)=0,05*131,5*18*10-3=0,12г
CaCl2 и Hg2(NO3)2 реагируют в эквивалентном количестве, т.е. в реакцию вступает одинаковое количество молей этих веществ.
n(Hg2(NO3)2)= m(Hg2(NO3)2)/ M(Hg2(NO3)2)=0,12/586=0,2*10-3 моль. Значит, n(CaCl2) также равно 0,2*10-3 моль.
m (CaCl2)=n(CaCl2)*M(CaCl2);
m (CaCl2)= 0,2*10-3*111=0,022 г – это масса в 200 мл, следовательно масса в литре раствора составит: 0,022*5=0,11г
Т.е. концентрация хлорида кальция в растворе составит 0,11 г/л.
Ответ: с(CaCl2)= 0,11 г/л.
4. При фотометрировании раствора сульфосалицилатного комплекса железа получили относительную оптическую плотность раствора 0,55. Раствор сравнения содержал 0,0288 мг железа в 25 мл. Толщина поглощающего слоя 2 см. Определить концентрацию и массу железа в 100 мл анализируемого раствора, если ε комплекса в этих условиях равен 3000 л/(моль·см).
D=eDcl, где e - коэффициент пропорциональности, который не зависит от концентрации, а зависит только от природы растворенного вещества;
Dc=с1-с0 – разность между концентрциями исследуемого раствора и эталонного;
l – толщина поглощающего слоя.
Отсюда, с1=D/(e*l)+ с0
Для начала необходимо вычислить с0.
Если в 25мл – 0,0288 мг, то
в 1000мл - х. х=1000*0,0288/25=1,15мг=1,15*10-3г,следовательно с0=1,15*10-3г/л
Однако необходима концентрация в моль/л.
n0=m0/M0; n=(1.15*10-3г)/56=0,02*10-3моль, следовательно с0=0,02*10-3моль/л.
с1=(0,55/(3000*2))+0,02*10-3=1,1*10-3моль/л
m1=n1*M1; m1=1.1*10-3моль/л*56=61,6мг
Т.е. с1=61,6мг/л
В 100 мл раствора масса железа будет в 10 раз меньше, т.е. будет составлять 61,6/10=6,16 мг.
Ответ: с1=61,6мг/л; m в 100 мл =6,16 мг.
5. Газовая хроматография. Сущность метода. Параметры удерживания.
Газовой хроматографией называется хроматографический метод, в котором в качестве подвижной фазы применяется газ или пар. В свою очередь газовая хроматография может быть разделена на газо-адсорбционную (газо-твердую) и газо-жидкостную. В первом случае неподвижной фазой служит твердое вещество — адсорбент, во втором — жидкость, распределенная тонким слоем по поверхности какого-либо твердого носителя (зерненого материала, стенок колонки).
Газоадсорбционная хроматография
Особенность метода газоадсорбционной хроматографии (ГАХ) в том, что в качестве неподвижной фазы применяют адсорбенты с высокой удельной поверхностью (10—1000 м2г-1), и распределение веществ между неподвижной и подвижной фазами определяется процессом адсорбции. Адсорбция молекул из газовой фазы, т.е. концентрированно их на поверхности раздела твердой и газообразной фаз, происходит за счет межмолекулярных взаимодействий (дисперсионных, ориентационных, индукционных), имеющих электростатическую природу. Возможно, образование водородной связи, причем вклад этого вида взаимодействия в удерживаемые объемы значительно уменьшается с ростом температуры.
Для аналитической практики важно, чтобы при постоянной температуре количество адсорбированного вещества на поверхности Сs было пропорционально концентрации этого вещества в газовой фазе Сm:
Cs = кcm,,
т.е. чтобы распределение происходило в соответствии с линейной изотермой адсорбции (к — константа). В этом случае каждый компонент перемещается вдоль колонки с постоянной скоростью, не зависящей от его концентрации. Разделение веществ обусловлено различной скоростью их перемещения. Поэтому в ГАХ чрезвычайно важен выбор адсорбента, площадь и природа поверхности которого обусловливают селективность (разделение) при заданной температуре.
С повышением температуры уменьшаются теплота адсорбции DH/T, от которой зависит удерживание, и соответственно tR . Это используют в практике анализа. Если разделяют соединения, сильно различающиеся по летучести при постоянной температуре, то низкокипящие вещества элюируются быстро, высококипящие имеют большее время удерживания, их пики на хроматограмме будут ниже и шире, анализ занимает много времени. Если же в процессе хроматографирования повышать температуру колонки с постоянной скоростью (программирование температуры), то близкие по ширине пики на хроматограмме будут располагаться равномерно.
В качестве адсорбентов для ГАХ в основном используют активные угли, силикагели, пористое стекло, оксид алюминия. Неоднородностью поверхности активных адсорбентов обусловлены основные недостатки метода ГАХ и невозможность определения сильно адсорбирующихся полярных молекул. Однако на геометрически и химически однородных макропористых адсорбентах можно проводить анализ смесей сильнополярных веществ. В последние годы выпускают адсорбенты с более или менее однородной поверхностью, такие, как пористые полимеры, макропористые силикагели (силохром, порасил, сферосил), пористые стекла, цеолиты.
Наиболее широко метод газоадсорбционной хроматографии применяют для анализа смесей газов и низкокипящих углеводородов, не содержащих активных функциональных групп. На молекулярных ситах — высокопористых природных или синтетических кристаллических материалах, все поры которых имеют примерно одинаковые размеры (0,4—1,5 нм), — можно разделить изотопы водорода. Сорбенты, называемые порапаками, используют для разделения гидридов металлов (Ge, As, Sn, Sb) (см. рис. 8.15). Метод ГАХ на колонках с пористыми полимерными сорбентами или углеродными молекулярными ситами самый быстрый и удобный способ определения воды в неорганических и органических материалах.
Газожидкостная хроматография
В аналитической практике чаще используют метод газожидкостной хроматографии (ГЖХ). Это связано с чрезвычайным разнообразием жидких неподвижных фаз, что облегчает выбор селективной для данного анализа фазы, с линейностью изотермы распределения в более широкой области концентраций, что позволяет работать с большими пробами, и с легкостью получения воспроизводимых по эффективности колонок.
Механизм распределения компонентов между носителем и неподвижной жидкой фазой основан на растворении их в жидкой фазе. Селективность зависит от двух факторов: упругости пара определяемого вещества и его коэффициента активности в жидкой фазе. По закону Рауля, при растворении упругость пара вещества над раствором pi прямо пропорциональна его коэффициенту активности g молярной доле Ni в растворе и давлению паров чистого вещества Р°i при данной температуре:
pi = Ni Р°i
Поскольку концентрация i-го компонента в равновесной паровой фазе определяется его парциальным давлением, можно принять что
Pi ~ cm, а Ni ~ cs. Тогда
![]()
а коэффициент
селективности ![]()
Таким образом, чем ниже температура кипения вещества (чем больше P0i), тем слабее удерживается оно в хроматографической колонке.
Если же температуры кипения веществ одинаковы, то для их разделения используют различия во взаимодействии с неподвижной жидкой фазой: чем сильнее взаимодействие, тем меньше коэффициент активности и больше удерживание.
Для обеспечения селективности колонки важно правильно выбрать неподвижную жидкую фазу. Эта фаза должна быть хорошим растворителем для компонентов смеси (если растворимость мала, компоненты выходят из колонки очень быстро), нелетучей (чтобы не испарялась при рабочей температуре колонки), химически инертной, должна обладать небольшой вязкостью (иначе замедляется процесс диффузии) и при нанесении на носитель образовывать равномерную пленку, прочно с ним связанную. Разделительная способность неподвижной фазы для компонентов данной пробы должна быть максимальной.
Различают жидкие фазы трех типов: неполярные (насыщенные углеводороды и др.), умеренно полярные (сложные эфиры, нитрилы и др.) и полярные (полигликоли, гидроксиламииы и др.).
Зная свойства неподвижной жидкой фазы и природу разделяемых веществ, например класс, строение, можно достаточно быстро подобрать подходящую для разделения данной смеси селективную жидкую фазу. При этом следует учитывать, что время удерживания компонентов будет приемлемым для анализа, если полярности стационарной фазы и вещества анализируемой пробы близки. Для растворенных веществ с близкой полярностью порядок элюирования обычно коррелирует с температурами кипения, и если разница температур достаточно велика, возможно полное разделение. Для разделения близко - кипящих веществ разной полярности используют стационарную фазу, селективно - удерживающую один или несколько компонентов вследствие диполь - дипольного взаимодействия. С увеличением полярности жидкой фазы время удерживания полярных соединений возрастает.