Реферат: Полимеразная цепная реакция и электрофорез
Реферат на тему:
ПЦР-реакция и электрофорез
2009
ПЦР-реакция
Может возникнуть необходимость увеличить количество индивидуальной кДНК до ее введения в плазмиду с помощью какого-то процесса ее воспроизводства. Такой процесс разработан и широко применяется не только для решения этой частной задачи, но и во всех случаях, когда необходимо умножить количество определенных фрагментов ДНК. Например, фрагментов, содержащих изучаемый ген и его регуляторное окружение.
Процесс этот получил название ПЦР-реакция, что расшифровывается как полимеразная цепная реакция. (В английском названии PCR — polymerase chain reaction.) Невольно напрашивается сопоставление с реакцией атомного взрыва. И, пожалуй, оно не лишено смысла — нарастание количества нужной ДНК происходит чрезвычайно быстро, даже бурно.
Рассмотрим эту реакцию для общего случая — наработки множества копий некоего фрагмента ДНК, содержащего в какой-то своей части интересующую нас последовательность пар оснований. Итак, допустим, что нам известна последовательность нуклеотидов на достаточно большом протяжении некой ДНК и есть основания предполагать, что внутри этой последовательности лежит интересующий нас участок. Рассмотрим серию операций, ведущих к его выделению и умножению.
Сначала в местах, лежащих близко, но заведомо за пределами интересующего нас участка ДНК, выберем две последовательности — по 20 пар нуклеотидов, лежащих по обе стороны от этого участка. Исходя из этих последовательностей синтезируем химически два однонитевых праймера на основе дезоксирибонуклеотидов. Первый — комплементарно к условно «первой» нити ДНК с ее Законна, второй — комплементарно ко «второй» нити с ее Законна. Очевидно, что праймеры будут разные.
Теперь добавим в буфер, где растворено малое количество исходной ДНК оба праймера в большом избытке, нагреем смесь до температуры 94°, а потом быстро охладим до 50°. ДНК денатурируется при 94°, ее нити расходятся. При 50° оба праймера гиб-ридизируются с выбранными для них участками однонитевых ДНК. Это произойдет быстро, так как праймеры имеются в избытке и, кроме того, вследствие своей малости они подвижны и легко «найдут» свои посадочные места. Ренатурация ДНК происходит медленно. За те 2 минуты, что будет продолжаться этот этап, ДНК практически не ренатурируется, а праймеры успеют надежно гибридизоваться с комплементарными для них участками обеих нитей. Такая ситуация отражена на рисунке 35 (1). Праймеры, как обычно, садятся с 3"-конца матричной нити ДНК и направляют движение будущей ДНК-полимеразы к ее 5'-концу. Это направление указано стрелками. Для удобства описания дальнейших событий, присвоим праймерам названия «левый» (стрелка зачернена) и «правый» (стрелка не зачернена).
Теперь быстро поднимем температуру смеси до 72 °С. при этой температуре нити ДНК заведомо не сойдутся, а праймеры еще удержатся на своих местах. Внесем в раствор ДНК-полимеразу. Вообще-то говоря, она там была с самого начала!
Но что это за фермент, который не денатурируется при 94°, а при 72° сейчас начнет вести комплементарный синтез ДНК? К счастью, такая ДНК-полимераза существует. Ее называют «Taq ДНК-полимераза» и выделяют из очень термофильных бактерий «Thermus aquaticus», прекрасно размножающихся при температуре 85 °С.
Итак, переходим к этапу 2, где изображен процесс матричного синтеза комплементарной нити ДНК, начинающийся от праймера и продолжающийся без других ограничений (что отмечено стрелой), кроме ограничения длительности этого этапа (3 минуты). Мы будем для простоты рисунка рассматривать события, начинающиеся с копирования только одной нити, хотя, конечно, будут копироваться обе. Они совершенно равноправны, и в заключение нашего анализа надо будет просто удвоить полученный результат. Через 3 минуты оканчивается 2-й этап и температура снова скачком поднимается до 94°, а еще через одну минуту быстро снижается до 50°. Эта ситуация отражена на этапе 3. При 94° новосинтезированная нить ДНК отделилась от материнской нити. (Последнюю, для ясности, я здесь и всюду далее изображаю жирной линией.) В составе новосинтезированной копии я больше не изображаю «левый» праймер, поскольку он был комплементарен материнской нити ДНК и потому вместе с участком, синтезированным ДНК-полимеразой, вошел в состав копии. Зато на эту копию с ее 3'-конца при 50° на предназначенный для него участок (ведь копия тождественна «второй» материнской нити) уже сел «правый» праймер. На освободившуюся материнскую нить с ее З*-конца тоже сел праймер — «левый», но, конечно, не тот, что ушел с копией, а другой, точно такой же. Благо праймеры имеются в избытке.
На этапе 4 (при 72°) показаны два новых комплементарных синтеза. Тот, что идет по материнской нити, по-прежнему, пространственно не ограничен. А вот тот синтез, что начинается от правого праймера, сидящего на новосинтезированной копии окончится там, где в этой копии «спрятан» весь бывший «левый» праймер — ведь с него эта копия начиналась. В результате здесь впервые появляется выбранный для умножения участок ДНК, ограниченный двумя праймерами, включая и их самих. Это хорошо видно на этапе 5, когда после нагрева до 94° все двойные нити разошлись и на рисунке оказываются уже 4 одинарных нити, на которые, туда «где им положено» село 4 праймера (два «левых» и два «правых»). Этап 6 — синтез 4-х копий, начинающихся от этих праймеров. В трех случаях из четырех он ограничен длиной нужного отрезка ДНК. (Материнская нить здесь, как и всюду дальше копируется без ограничения справа.) Вместе с образованным на 4-м этапе и уже обрезанным с обеих сторон участком мы получаем 4 фрагмента исходной ДНК нужного размера. Это хорошо видно на этапе 7, где все четыре пары нитей ДНК разошлись и на них уже сидят 8 праймеров...
Если у читателя хватило терпения разобраться во всей этой «механике», то он согласится, что после синтеза на этапе 8 получится уже 11 отрезков ДНК нужной длины. Заметим попутно, что хотя мы рассматриваем «потомство» одной только «первой» материнской нити, среди полученных отрезков будут копии участков как «первой», так и «второй» материнской нити, поскольку мы уже не один раз вели комплементарный матричный синтез.
Проследим теперь закономерность, отраженную в цифрах, стоящих справа от рисунка, около изображений 3-го, 5-го, 7-го и 8-го этапов. Перед скобкой каждый раз стоит число одиночных нитей ДНК после нагревания до 94°. Легко заметить, что оно неизменно удваивается. Что и следовало ожидать, поскольку на каждом из предыдущих четных этапов все имеющиеся в наличии нити ДНК так или иначе копируются.
Но вот что может показаться неожиданным, и в чем состоит вся суть ПЦР-реакции. Число фрагментов нужного размера, указанное в скобках, нарастает несравненно быстрее: 0-1-4—11 штук. Так будет и далее. Каждый укороченный отрезок будет копироваться в том же размере. И число их будет непрерывно пополняться за счет не сразу укороченных отрезков ДНК. Через 30 циклов, подобных рассмотренным (а каждый цикл — это два этапа) количество нитей ДНК достигнет огромной цифры. Притом практически все они уже будут нужной длины — и выделение фрагмента, и его умножение состоялось! Вспомним, что у нас исходно было две нити. Таким образом написанное число надо удвоить.
Что это означает не в штуках, а в весовых единицах? Можно подсчитать, что если имелось изначально всего 10 молекул ДНК, длиной в 1000 пар оснований каждая, то в результате 30-ти циклов ПЦР-реакции должно получиться около 2-х микрограммов необходимого генетического материала. Для современных методов исследования это весьма значительное количество.
На самом деле в таких подсчетах конечный выход ДНК получится значительно завышенным, потому что Taq ДНК-полимера-за изнашивается, а после 30 циклов и вовсе перестает «работать». Но ведь можно внести новую порцию фермента и запустить еще 30 циклов. (Замечу попутно, что Taq ДНК-полимераза не очень «строга». После 30 циклов в среднем 1 нуклеотид из 400 оказывается включенным ошибочно.)
Разумеется, все эти циклы осуществляются не вручную, а в специальном приборе, от которого, впрочем, требуется не многое. Только очень быстро по обозначенной выше программе менять температуру весьма малого объема жидкости (защищенной от испарения тонким слоем минерального масла). Что же касается продолжительности 30-ти циклов, то даже, если учесть, что длительность синтеза приходится по указанной выше причине постепенно увеличивать от 3-х до 10-ти минут, то на один цикл прибор будет затрачивать в среднем 12 минут. А на 30 циклов — 6 часов.
От экспериментатора требуется только правильно составить рабочую смесь. Разумеется, если праймеры уже выбраны и синтезированы в достаточном количестве. Taq ДНК-полимераза и нуклеозидтрифосфаты имеются в продаже. Наработку большого количества определенного гена при помощи ПЦР-реакции часто называют «клонированном» этого гена. Описанную здесь ПЦР-реакцию с двумя праймерами иногда именуют «симметричной», в отличие от другой тоже ПЦР-реакции, но с одним начальным праймером, которую называют «ассиметричной».
ПЦР-реакцию надо включить в описанную там последовательность операций между получением кДНК и включением ДНК в плазмиду. В этом случае последовательность 21-го нуклео-тида для праймеров придется выбирать не свободно, а точно по концам гена, кодирующего наш белок. Эти оба конца можно установить, как это было описано с помощью ЧИП-метода. Для этого даже не надо знать всю аминокислотную последовательность белка, а только концевые участки — по 7 аминокислот с каждого конца. (Благо, как упоминалось, секвенирование белка теперь можно начинать с любого конца.) При синтезе концевого праймера надо только добавить концевой кодон УГА, который не транскрибируется в иРНК. Кроме того к «наружным» концам обоих праймеров имеет смысл уже на этом этапе добавить небольшие последовательности нуклеотидов, которые, не будучи комплементарны ни к какому участку гена, не будут и гибридизоваться. Но могут образовать два «липких» конца для последующего включения размноженной кДНК в разрезанные плазмиды. На рис. 35 эти дополнительные последовательности изображены в виде «хвостиков» у праймеров. Напомню, что эта размноженная кДНК нам потребовалась для того, чтобы добиться достаточно эффективного включения содержащих ее плазмид в бактерию, которая, размножаясь, будет нарабатывать в большом количестве нужный нам белок.
Весь полный комплекс мер по умножению количества индивидуального белка иной раз оказывается столь эффективным, что чужеродный белок в цитоплазме бактерий-рециплиентов появляется в ходе их размножения в виде гранул. Он ведь чужой — и потому не расходуется в самой бактерии. После лизиса клеток гранулы можно собрать простым центрифугированием. Для их диссоциации приходится использовать обработку осадка щелочью, детергентами и мочевиной. Белок денатурируется. Для его ренатурации приходится прибегать к разбавлению, диализу от денатурирующих добавок, изменению рН и ионной силы среды.
Подлинность наработанного белка проверяют по ферментативной активности, если он должен таковой обладать. Или же по электрофорезу — сравнением с контрольным препаратом. Но надежнее — одним из иммунологических методов контроля, с которыми мы познакомимся в свое время.
2. Электрофорез
Метод электрофореза таит в себе массу «подводных камней», отчего слепое копирование описанных в научной литературе примеров его использования приводит, как правило, к плачевным результатам. Поэтому попробуем разобраться в физических основах метода поглубже..
2.1 Введение в метод электрофореза
Представим себе два емких сосуда, соединенных между собой тонкой и длинной стеклянной трубочкой, наподобие буквы Н. Пусть сосуды и трубочка заполнены слабым раствором поваренной соли. В сосуды опустим электроды — проволочки, соединенные с клеммами источника постоянного напряжения. К примеру, пусть проволочка из правого сосуда присоединена к клемме «-», а из левого — к клемме «+». Это будут, соответственно, наши катод и анод. Включим напряжение. Миллиамперметр источника покажет, что в замкнутой цепи протекает некий ток. Он течет через солевой раствор, в частности и вдоль трубочки. Она-то нас и интересует. Вдумаемся в то, что в ней будет происходить. Никаких других растворенных веществ в трубочке нет. Электрический ток обусловлен исключительно движением двух ионов — отрицательными ионами Сl и положительными Na+. Первые движутся влево, к аноду, вторые — вправо, к катоду.
Следует ли опасаться, что запас ионов С1- и Na+ в трубочке со временем истощится? Нет. Потому что из резервуара катода в трубочку будут входить ионы Сl-, а из резервуара анода — ионы Na+, поддерживая неизменной концентрацию обоих ионов в ней. Во всяком случае так будет продолжаться до тех пор, пока не исчерпаются или хотя бы существенно изменятся запасы этих ионов в самих резервуарах. Мы до этого доводить не будем.
Зададимся теперь наивным вопросом: а что заставляет какой-нибудь конкретный ион (пусть С1-), находящийся в трубочке, двигаться влево по направлению к аноду? Ответ очевидный _ электрическое поле. А конкретнее?
Раз по трубочке течет электрический ток, значит она играет роль проводника и, следовательно, на нее подается определенное «напряжение». Ну а откуда, спросим себя, некий индивидуальный ион С1-, находящийся в середине трубки «знает», что к ее концам приложено напряжение? Но коль скоро к концам любого проводника приложено напряжение, то в этом проводнике на всей его длине немедленно образуется электрическое поле. Оно будет тем интенсивнее (сильнее), чем больше напряжение и короче трубочка. Величину интенсивности электрического поля в любой точке однородного проводника определяют как Е = V/1, где V _ напряжение, которое подается на проводник, а 1 — его длина. Эту величину именуют «напряженностью» электрического поля в данной точке проводника. Единицей напряженности, очевидно, является В/см.
Величину напряженности поля и «чувствует» любой ион, находящийся в этой точке, она заставляет его двигаться к соответствующему электроду. В однородном проводнике напряженность поля одинакова в любой точке. Подчеркнем, что напряженность поля не зависит (практически) от находящихся в трубочке в малых количествах веществ, хотя бы тоже ионов. Ее определяют основные носители тока, данном случае ионы С1- и Na+. Но сами эти «посторонние» вещества, если они тоже ионы, будут в полной мере испытывать воздействие электрического поля.
Сила, действующая на любой ион равна произведению величины его заряда на напряженность поля. В нашей трубочке она постоянна и мы могли бы ожидать на основе законов механики, что все ионы движутся равноускоренно. Этого не происходит из-за сопротивления, которое оказывает такому движению окружающая среда, в данном случае вода. В результате каждый заряженный ион будет «пробиваться», мигрировать к своему аноду довольно медленно и с постоянной скоростью, поскольку сила трения увеличивается с увеличением скорости миграции до тех пор, пока она не сравняется с силой, влекущей ион к его электроду. У разных ионов эта скорость может быть различной, поскольку сила трения зависит еще и от размера иона. Вообще, скорость миграции иона будет тем больше, чем больше его заряд и напряженность электрического поля и чем меньше размер иона. Ионы С1- и Na+ примерно одинаковой величины и потому мигрируют в разных направлениях, но с примерно одинаковой скоростью. Совсем другое дело, если бы вместо NaCI мы растворили в воде знакомый нам Трис-НСl буфер. Нам известно, что при исходной концентрации Триса =0,1 M и добавлении в его водный раствор НС1 до 0,05 М (при рН8) примерно половина молекул Tpиca превращаются в ионы Трис+ и точно такое же количество в растворе появляется ионов С1-. Ион Трис+ в 3 раза тяжелее и в несколько раз крупнее, чем ион С1-. Соответственно он будет медленнее мигрировать в электрическом поле. А следовательно, основными носителями тока в этом случае будут ионы С1- (хотя свой небольшой вклад дадут и ионы Трис+). Более того, если бы мы вообще каким-либо образом «перегородили дорогу» ионам Трис+, то весь ток переносили бы только ионы С1- (подобно электронам в металлическом проводнике). Напряженность поля установится и прочие заряженные вещества смогут двигаться в этом поле в соответствии со своими зарядами и размерами. Можно сказать, что напряженность поля определяется основными носителями заряда — они создают реальное сопротивление проводника, следовательно и величину напряжения от источника тока, которое приходится на его долю. А тем самым и напряженность поля, если длина проводника (трубочки) неизменна и он однороден.
2.2 Электрофорез белков (общие положения)
Теперь предположим, что с помощью пипетки с отогнутым концом мы ввели в начало трубочки, заполненной Трис-НСl буфером смесь различных молекул белка. Мы знаем, что каждая молекула белка может быть положительно или отрицательно заряжена. Этот заряд определяется как сумма электрических зарядов боковых групп аминокислот: основных (лизин, аргинин, гистидин) и кислых (аспарагиновая и глутаминовая кислоты), лежащих на поверхности белка. Боковые группы, спрятанные внутри белковой глобулы, своего вклада в суммарный заряд белка не дают, так как не соприкасаются с водой и потому не ионизированы.
Начнем рассмотрение с наиболее распространенных кислых белков, т. е. таких, чьи суммарные заряды отрицательны, хотя степень их кислотности, наверное, будет разной — она зависит от соотношения числа заряженных групп разных знаков на поверхности.
Под действием того же самого электрического поля такие белки, подобно иону Сl- будут мигрировать в сторону анода. Они тоже будут испытывать сопротивление своему движению и наверняка большее, чем ион С1- в силу своих размеров и необходимости раздвигать лежащие на их пути молекулы воды.
Когда электрические силы, влекущие молекулы белков к аноду, станут равными силам сопротивления (последние тем больше, чем быстрее движение), скорости миграции белков тоже станут постоянными и, скорее всего, разными у различных белков. Эти скорости будут пропорциональны величинам суммарных зарядов каждого из белков и, разумеется, напряженности общего для них всех электрического поля. Напряженность поля зависит от электропроводности буфера, т. е. концентрации основных носителей тока (в нашем примере — ионов С1-). Присутствие белков, несмотря на их заряды, существенного влияния на электропроводность раствора иметь не должно ввиду малости их количества. Зато величины зарядов белков (а значит и скорости их миграции) будут весьма существенно зависеть не только от их природы, но и от величины рН раствора, создаваемого буфером. Собственно говоря, именно ради того, чтобы иметь возможность управлять зарядами белков, мы заменили в нашем растворе соль на буфер. Создаваемое им рН надо выбрать не так, чтобы все белки получили максимально возможный отрицательный заряд (например, в сильно щелочном буфере). Это невыгодно. Все белки будут двигаться с максимальными скоростями, не сильно отличающимися друг от друга. Выгодно выбрать умеренно щелочную среду и сыграть, таким образом, на различии соотношений основных и кислых боковых групп аминокислот на поверхности разных белков. Иными словами, добиваться максимального различия сил, действующих на разные белки — тогда на своем пути вдоль трубки они разойдутся наиболее явным образом. (Обычно для кислых белков выбирают величину рН8-8,5, а для щелочных, — гистонов, белков рибосом, — используют буферы с рН4—5. Эти последние белки будут мигрировать к катоду.)
О том, как мы будем обнаруживать и трактовать расхождение белков в геле речь впереди. А пока продолжим разговор о выборе буфера. Выше шла речь о выборе его рН. А как выбрать концентрацию буфера? Чем она выше, тем больше, как говорят, «емкость» буфера — его способность удерживать рН раствора от резких изменений при внесении в него кислот и щелочей. Но ведь мы, как будто, не собираемся их вносить? Оказывается вносим! С самими белками.
Вернемся теперь к белкам в электрофорезе. Мы их вносили в трубочку, очевидно, уже растворенными в выбранном для них буфере. Раствор этот был более или менее разбавленным. В ходе электрофореза белки, как мы увидим ниже, будут собираться в узкие зоны, где концентрация каждого из них окажется гораздо выше чем изначально. Вот там-то емкости буфера может не хватить, рН раствора может измениться. За ним изменится и суммарный заряд белка, а следовательно и скорость миграции белковой зоны. Устранить эту опасность, на первый взгляд, легко. Достаточно в несколько раз увеличить концентрацию буфера. Но это будет означать, к примеру для Трис-НСl буфера, такое же увеличение концентрации ионов С1-. А значит и увеличение силы тока и, как следствие, усиление разогрева жидкости в трубочке. В результате чего возникнут искажения картины разделения белков. Хотя бы потому, что температура жидкости в центре трубочки будет выше, чем у ее стенок. Но ведь можно уменьшить напряжение на клеммах источника. Но это приведет к падению напряженности поля и соответствующему замедлению миграции белков. Что нежелательно ввиду диффузного размытия областей (полос) миграции. В порядке компромисса выбирают обычно молярность буфера в пределах 0,1-0,2М.
Нежелательная ситуация может легко возникнуть и в том случае, когда рН рабочего буфера выбрана близ границы буферной области (см. гл. 2, § 1). Здесь буферная емкость мала по определению. И потому вышеописанный эффект изменения рН в зоне концентрации белка может быть особенно опасен.
Вполне возможно, что столкнувшись с такими трудностями экспериментатору придется отказаться от первоначально выбранного буфера и заменить его на другой, с менее подвижными ионами.
Я не собираюсь приводить здесь практические рецепты. Мне только хотелось показать, что выбор буфера для электрофореза дело тонкое, требующее вдумчивой оценки данной конкретной ситуации.
Теперь обратимся к другой стороне проблемы. До сих пор мы рассматривали только равновесие электрических сил, действующих на белки и сил трения в жидкости. Размер частиц в этом случае может сказываться двояко. С одной стороны будет увеличиваться сила трения в водной среде, но, с другой стороны, может увеличиться и суммарный электрический заряд на поверхности белковой глобулы. Попробуем различие размеров разных белков использовать более определенным образом. Для этого создадим помимо трения о жидкость еще и дополнительное трение — напрямую связанное с размерами мигрирующих молекул белка. Создадим искусственные преграды для миграции в виде пространственной сетки, ячейки которой будут соизмеримы с размерами белков. Эта сетка должна быть образована волокнами, которые надежно смачивает вода, коль скоро мы будем работать в водных растворах. В этом случае буфер будет заполнять ячейки сетки целиком. Хорошо бы, чтобы в нашем распоряжении была возможность выбирать размеры ячеек или пор этой сетки — в зависимости от диапазона размеров белков, которые мы собираемся разделять. Очевидно, что все молекулы белков должны иметь возможность проходить через поры, ни в коем случае не застревать в них. Но проходя, они должны постоянно сталкиваться с образующими поры нитями, что будет тормозить их миграцию в электрическом поле. И чем крупнее белки, тем чаще им придется испытывать эти столкновения, тем «труднее« и медленнее они будут мигрировать. Самые мелкие белки, напротив, будут проходить через поры едва касаясь их нитей. Скорость миграции для белков различной величины, опять-таки, установится постоянной, поскольку статистически постоянным будет эффект столкновения с нитями — он зависит только от соотношения размеров молекул белка и пор. Мы получаем мощное средство разделения белков по их размерам.
Таким образом, выбирая пористость сетки и оценивая результаты разделения белков придется принимать во внимание не только их массу, но и конфигурацию. Мало того, придется учитывать и жесткость (плотность упаковки) белковой молекулы. Рыхлые глобулярные (и особенно фибриллярные) белки могут деформироваться при взаимодействии с сеткой и тем самым облегчать себе миграцию между ее нитями.
Но что же это за сетка, которую мы хотим создать? И как это сделать? Скорее всего это будет гель, подобный пищевому желе, которое получают очень небольшой добавкой желатина к фруктовому соку, или нечто похожее на уже знакомый нам агар. Прежде, чем поговорить об этом геле подробнее, отметим, что благодаря силам смачивания жидкость из геля с достаточно мелкими порами не будет вытекать, даже если она составит 95% его массы. А это значит, что заполнив нашу трубочку гелем, мы можем поставить ее вертикально, как это показано на рис. 36. Кстати сказать, так и выглядели первые приборчики для электрофореза.
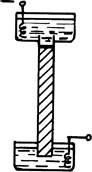
Рис. 36
Вертикальное расположение трубочки с гелем сразу обнаруживает одно существенное преимущество. Теперь препарат исходной смеси белков можно наносить тонким ровным слоем на верхнюю поверхность геля. В результате чего и разделяющиеся в ходе миграции в электрическом поле белки будут двигаться вниз в виде тонких дисков. (Каждый со своей скоростью, постепенно располагаясь на всей длине трубочки.)
Разумеется, буферы верхнего и нижнего резервуаров должны смачивать оба торца геля в трубочке. Поэтому гель не должен заполнять трубочку до самого верху, оставляя место для нанесения препарата. В исходную белковую смесь (тоже растворенную в буфере) можно добавить 5-10% сахарозы или глицерина. В таком виде ее можно вносить в трубочку пипеткой с оттянутым полиэтиленовым наконечником, осторожно подслаивая под буфер, находящийся в трубочке.
В ходе электрофореза зоны растворенных белков остаются невидимыми. Для наблюдения за процессом разделения в исходный препарат добавляют 0,01% красителя, молекулы которого несут на себе электрический заряд того же знака, что и фракционируемые белки, но не взаимодействуют с ними. Краситель в электрическом поле перемещается вдоль трубочки в виде окрашенной полоски. Его подбирают таким образом, чтобы скорость его миграции была немного больше, чем скорость наиболее подвижных молекул белка. Когда окрашенная полоска доходит до конца трубочки электрофорез прекращают. В качестве отрицательно заряженного красителя широко используют «Бромфеноловый синий». Для электрофореза щелочных, положительно заряженных белков — «Метиловый зеленый» или «Пиронин».
После окончания электрофореза столбик геля извлекают из трубочки, фиксируют в конечном положении миграции и белки окрашивают — прямо в геле. К методам такой фиксации и окраски мы еще вернемся, а сейчас надо будет познакомиться с природой самого геля, используемого для фракционирования белков.
Литература
1 Курашвили Л.В., Ковалев К.В. Атерогенные липопротеиды у больных с абдоминальной патологией. Научно-практичеекая конференция, посвященная 140-летию областной больницы им.Н.Н. Бурденко и в честь 110-летия со дня рождения академика Н.Н. Бурденко. Тез.докладов. - Пенза. 1986. - С.101-102.
2 Курашвили Л.В. , Устинова Т.И. Лабораторные тесты в диагностике гипоксических соотояний. Научные чтения в часть памяти академика Н.Н. Бурденко). - Пенза, 1988. С.148-150.
3 Курашвили Л.В., Волков А.С., Прокаева П.А. Коэффициент атерогевности и холестерин в диагностике нарушений липидного обмена. VI Научные чтения памяти академика Н.Н. Бурденко. - Пенза, 1988. - С.165-166.
4 Курашвили Л.В., Савченко Р.П. Метаболизм липидов у больных в терминальной стадии хронической почечной недостаточности // Казанский медицинский журнал. - 1990. - Т.XXI. - N 5. - С.338-340.