Реферат: Искусственный синтез олигонуклеотидов
Реферат на тему:
Искусственный синтез олигонуклеотидов
2009
1. Искусственный синтез олигонуклеотидов
Искусственный химический синтез заданной последовательности рибо- или дезоксирибонуклеозидов (типа РНК и ДНК).
Первый нуклеотид последовательности через его 5'-фосфатную группу химически закрепляют на твердой подложке, например, полистирола. Все потенциально химически активные группы и атомы его нуклеинового основания (аминогруппы, кислород) временно блокируются присоединением инертной «защиты». 3'-ОН группа сахара не блокируется.
Все нуклеотиды, подлежащие последовательному присоединению тоже заблаговременно защищаются не только по нуклеиновым основаниям, но и по 3'-ОН группе рибозы или дезоксирибозы. Активность фосфорной кислоты, присоединенной по 5'-положению сахара умеряется «навеской» хлорбензола по одному из ее кислородов. Таким образом, для реакции остается свободной только ОН-группа этой кислоты.
Теперь в реакционную среду вносят второй по заданному порядку нуклеотид последовательности — защищенный, как это было только что описано. В реакции между двумя ОН-группами отщепляется вода и устанавливается ковалентная фосфодиэфирная связь двух нуклеотидов. Затем снимается химическая защита с 3'-ОН группы только что присоединившегося второго нуклеотида и в реакционную среду вносят третий защищенный нуклеотид... И так далее. Разумеется, отщепление воды и образование фосфодиэфирной связи происходит не само собой, а в присутствии сложного химического катализатора реакции.
По окончании синтеза заданной цепочки олигонуклеотида все защиты снимаются и олигонуклеотид отделяется от твердой подложки.
Современные автоматические синтезаторы производят такую «сборку» олигонуклеотидов по заданной программе на длину до сотни нуклеиновых оснований.
Упомяну еще, что недавно найден способ наращивания искусственной цепи олигонуклеотидов с заменой химического катализатора на облучение этой цепи лучом лазера на каждой следующей ступени синтеза. Это упоминание вскоре нам пригодится.
Получив описанным способом отрезок одной нити нужного фрагмента ДНК, построение второй нити по нему, как по матрице, можно поручить ДНК-полимеразе.
1.1 Секвенаторы белков
Остается сказать несколько слов об еще одном автомате, предоставляющем в распоряжение исследователя необходимую для всего дальнейшего последовательность аминокислот в исходном белке. Первый вариант такого автомата был предложен еще в 1967 году Эрдманом и Беггом. Он настолько хорошо зарекомендовал себя, что в основных чертах сохранился до сих пор. Единственное, но не маловажное его улучшение состоит в том, что современные секвенаторы могут анализировать аминокислотную последовательность с обоих концов белковой молекулы (N-конца и С-конца), в то время, как цепь реакций, предложенных Эрдманом решала только первую из этих двух задач.
Упомянутая цепь химических реакций в деталях нам еще не доступна, поэтому я ограничусь общей словесной характеристикой ее 3-х этапов, обращая внимание на физическое состояние и характеристики реагентов.
На 1-м этапе некое сложное органическое вещество в водной, слегка щелочной среде присоединяется к исследуемому белку с его N-конца. На 2-м этапе оно отделяется от белка, «прихватив» с собой концевую аминокислоту. Этот этап приходится проводить в кислой безводной среде. В результате получается сложное и весьма гидрофобное производное концевой аминокислоты и укороченный на одно звено белок. Их можно разделить между собой переводя в такой органический растворитель, в котором укороченный белок выпадает в осадок, а модифицированная аминокислота в этом растворителе может быть выведена из прибора для ее дальнейшей (хроматографической) идентификации. .Перед началом следующего цикла осадок белка должен быть вновь растворен в водной, слегка щелочной среде.
Из сказанного ясно, что в ходе полного цикла реакции необходимо менять растворители, осаждать один из компонентов (белок), экстрагировать другой, промывать осадок, сушить его и снова растворять. Все эти операции по определенной программе удается осуществить с помощью быстро вращающейся (>1500 об/мин) стеклянной чашечки, которая служит реакционным сосудом (рис. 32).
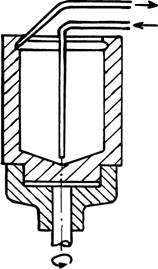
Рис. 32.
В нее вносят исходный препарат раствора белка. По центральной трубке на коническое дно чашечки один за другим подаются необходимые жидкие реагенты. Благодаря вращению они тонким слоем поднимаются по стенкам чашечки. Их количество и скорость вращения можно подобрать так, что слой не достигает канавки, выполненной в верхней части внутренней поверхности. Тонкая пленка прилегающей к стенке жидкости идеально подходит для экстракции из нее вещества в другую тонкую пленку. Ее образует не смешивающийся с предыдущей жидкостью органический растворитель, который поднимается по поверхности первой пленки. Здесь же осуществляется центрифугирование. На стенках чашечки оседают укороченные полипептидные цепи белка, в то время, как органический растворитель вымывает из реакционной смеси отщепившуюся гидрофобную производную очередной аминокислоты. Для этой цели растворитель подается в избытке, слой его поднимается до канавки, откуда через вторую трубочку раствор модифицированной аминокислоты поступает в коллектор фракций. Осажденный на стенки белок там же промывается и высушивается, а затем снова растворяется в слегка щелочной водной среде. Далее весь цикл повторяется...
За последующие годы прибор Эрдмана и Бегга был технически усовершенствован и переведен на компьютерный контроль, но первоначальный принцип его устройства сохранился.
2. Умножение количества индивидуального белка
Перспектива наработки большого количества индивидуального белка в качестве продукта деятельности вторгнувшегося с плазмидой в бактерию гена. Этот ген должен быть тем самым геном, который изначально кодировал синтез нужного нам белка. Только и всего! Но посмотрим, так ли просто получить этот ген, исходя только из наличия в нашем распоряжении самого белка? Будем решать эту задачу для белка животного происхождения. Здесь она в определенной мере сложнее ввиду сплайсинга иРНК и практически важнее. В ее решении явно просматриваются два этапа:
1) Получить индивидуальную иРНК, ответственную за синтез интересующего нас белка.
2) На базе этой иРНК создать двунитевую структуру ДНК, воспроизводящую если и не весь ген, то его «значащую» часть — ту совокупность последовательных участков, которые диктуют синтез соответствующих экзонов иРНК. Такой «рационально укороченный» ген можно встраивать в плазмиду. Он в бактерии-реципиенте обеспечит синтез полноценной иРНК, а вслед за ней и наработку нужного нам белка. Мне удобнее будет начать с этого второго этапа.
2.1 Воспроизведение «значащей» части гена по известной иРНК
Предположим, что мы располагаем достаточным количеством нашей индивидуальной иРНК. Рассмотрим последовательность операций, позволяющую произвести метаморфозу этой иРНК в соответствующую ей часть гена прямо в пробирке.
Нам известно, что иРНК животного происхождения на своем 3'-конце несет длинный «хвост» адениновых нуклеотидов (поли А).
1 этап. Добавим к раствору нашей иРНК в достаточном количестве олигонуклеотид длиной в десяток-другой дезоксириботи-мидинов (олиго дТ). Мы уже знаем, как его можно синтезировать. Олиго дТ может гибридизоваться с поли А. Создадим для этого оптимальные условия (буфер, концентрация соли, умеренно повышенная температура). Мы получим структуру, с которой начнется 2-й этап.
2 этап. Добавим в раствор нам еще не встречавшийся фермент — «обратную транскриптазу». В присутствии 4-х дезоксирибонуклеотидов этот фермент способен вести комплементарный синтез ДНК по матрице РНК в нормальном направлении от 3' к 5'-концу матрицы. Он тоже нуждается в праймере, каковым для него послужит уже сидящий на 3'-конце иРНК олиго дТ
3 этап. Обработаем полученный РНК-ДНК гибрид упомянутой ранее рибонуклеазой Н. Она как раз разрушает РНК, находящуюся в двунитевом гибридном комплексе с ДНК (см. там же). Однако поставим этот фермент в такие условия, чтобы наша иРНК была разрушена не до конца — остались небольшие ее участки.
4 этап. От этих участков, как от праймеров, начнет работать вносимая на этом этапе ДНК-полимераза I. Она, как нам известно, не только ведет матричный синтез ДНК, но и обладает S'-S* экзо-нуклеазной активностью. Благодаря этому она разрушает остатки иРНК и образует из синтезированных ею фрагментов, не без помощи все той же ДНК-лигазы, полноценную нить ДНК.
Полученную таким образом двухнитевую молекулу принято именовать «кДНК», имея в виду, что она является комплементарно выстроенным отображением исходной иРНК. Эта ДНК соответствует значащей последовательности нашего гена, поскольку все интроны (повторю это еще раз) были «вырезаны» уже при выходе исходной иРНК из ядра в цитоплазму. РНК-полимераза бактерии, которой придется транскрибировать плазмидную ДНК, на участке этой встроенной кДНК произведет полноценную иРНК для синтеза интересующего нас белка.
2.2 ЧИП-методика
Более простая часть задачи получения гена, когда в нашем распоряжении имеется нужная индивидуальная иРНК. Но как ее получить, если мы располагаем только некоторым количеством клеток животного происхождения, в которых синтезировался интересующий нас белок, и, следовательно, имеется соответствующая ему иРНК?
Очевидно начать надо с того, что отделить суммарную фракцию всех иРНК этих клеток от всех прочих РНК. Для этого достаточно закрепить на неподвижной твердой основе в достаточном количестве молекулы все того же олиго дТ и пропустить мимо них раствор всех выделенных РНК клеток. Это делается методом колоночной «аффинной» хроматографии, с которым мы в свое время будем знакомиться подробно. Но и так ясно, что нетрудно создать условия, при которых закрепленные неподвижно олиго дТ «выхватят» из протекающего мимо раствора суммарных РНК все молекулы иРНК путем гибридизации с их поли А «хвостами». Все прочие РНК спокойно протекут мимо. После чего любым достаточно мягким способом (нагреванием или действием слабой щелочи) можно будет разорвать водородные связи поли А — олиго дТ гибридов и получить в растворе суммарную фракцию всех иРНК.
Теперь дело за тем, чтобы из этой суммарной фракции выловить нужную нам индивидуальную иРНК, ответственную за синтез интересующего нас белка. Казалось бы это сделать не так уж сложно.
Мы можем без особого труда с помощью секвенатора белков установить аминокислотную последовательность (хотя бы и неполную) нашего белка. Далее, используя генетический код, установить некую последовательность нуклеотидов (не менее 20-ти), отвечающую определенной последовательности аминокислот (не менее семи) в нашем белке. Синтезировать эту последовательность, как описано выше, опять закрепить на неподвижной основе и пропустить мимо нее суммарный раствор всех иРНК. Благодаря гибридизации с достаточно протяженным участком своего гена (пусть и искусственно синтезированного) мы можем опять выловить теперь уже нашу индивидуальную иРНК. Ошибки тут быть не может. Вероятность того, что в двух разных иРНК окажутся одинаковые последовательности, комплементарные к одному и тому же участку гена длиной в 20 нуклеотидов практически равна нулю.
Как будто все просто, но... вспомним, что генетический код-то вырожденный . Нам неоткуда узнать как в данном конкретном случае была использована эта вырожденность. И это вдруг чрезвычайно усложняет задачу. Верхняя строчка обозначает последовательность из семи аминокислот, которую мы, предположим, выбрали из известной аминокислотной последовательности нашего белка. Выбрали далеко не самый тяжелый случай. Из семи аминокислот только двум отвечает по 4 кодона. Нет ни одной «шестикодонной» аминокислоты и даже фигурирует триптофан, у которого только один кодон. Даже и в этом случае для того, чтобы наверняка «поймать» нужную нам иРНК придется перепробовать все варианты. То есть синтезировать не одну последовательность из 21-го нуклеотида, а все возможные последовательности, исчерпывающие все допустимые комбинации использования разрешенных кодонов. Нетрудно подсчитать число их: 4-2'2'2-4'1-2 = 256.
Итак, надо будет синтезировать 256 21-членных олигонуклеотидов и 256 раз повторить опыты по гибридизации с суммарной фракцией всех иРНК. Это — огромная работа (хотя и пустячная по сравнению с некоторыми другими современными исследованиями, когда приходится ставить тысячи параллельных опытов по гибридизации).
Не будем пока думать о синтезе 256-ти олигонуклеотидов. Все-таки, их будет делать машина. Но 256 раз повторять опыты по гибридизации! Со всеми необходимыми промывками и контролями. Здесь крайне нужна автоматизация. А раз так, то соответствующий прибор должен появиться на рынке научной аппаратуры. И он появляется.
Представьте себе настольный прибор, ненамного больше обычного телевизора. На массивной, строго горизонтально установленной стальной плите имеется гнездо для точной фиксации некой «плашки». Последняя представляет собой прямоугольную пластинку из плексигласа размером 95 х 130 мм. В ней равномерно расположены 24 ряда лунок, по 16 в каждом ряду, вместимостью по 50 микролитров. Ряды и столбцы лунок пронумерованы. В них предварительно заливают по 10 микролитров смеси водного раствора мономеров, которые потом превратятся в пористый полиакриламидный гель, светочувствительный катализатор этого превращения и необходимые нам синтетические олигонуклеотиды. В данном примере 256 различных олигонуклеотидов надо будет внести в 256 лунок — согласно программе, указанной компьютером, который будет управлять всеми дальнейшими манипуляциями прибора.
В другом месте на той же массивной плите с помощью пружинящих упоров в определенные положения могут быть уложены до восьми предметных стекол от микроскопа. Верхняя поверхность этих стекол покрыта гидрофобной пленкой, несущей в себе активные химические вещества, способные под действием ультрафиолетового света связываться с полиакриламидным гелем. Кроме того на этой же пластине имеется два крупных колодца диаметром 2 см. Через один из них по команде от компьютера может прокачиваться дистиллированная вода. Во второй колодец заливают этанол. Наконец, еще в этой же плите проделано отверстие, ведущее в сушильную камеру, находящуюся под плитой.
На нижней поверхности второй массивной плиты, расположенной полуметром выше первой, смонтирована вся подвижная система. Она состоит из двух прецизионных ходовых винтов, вращением которых укрепленная на них «каретка» может быть доставлена в любое место над нижней плитой. Вращением винтов, естественно, тоже управляет компьютер.
На каретке смонтирована вертикальная стальная игла диаметром 0,3 мм с плоским нижним торцом. К ней установлено на стойке стальное кольцо диаметром в 1 мм. Игла может опускаться и подниматься — вместе с кольцом или отдельно от него так, что ее торец пересекает плоскость кольца.
По заданной программе механический привод за 1 секунду переносит иглу и кольцо в положение точно над очередной лункой плашки. Торец иглы во время этого переноса поднимается выше кольца. Затем кольцо мгновенно опускается в лунку. Благодаря малому диаметру кольца жидкость образует в его плоскости устойчивую пленку. Затем, также за 1 секунду, каретка переносит иглу с кольцом в заданное положение над одним из предметных стекол — с точностью в ±10 микрон! Сухой стержень иглы опускается, пронизывает пленку, смачивается и с легким ударом (пружинка!) касается своим торцом стекла. Жидкость с него стряхивается. На поверхности стекла образуется крошечная капля, диаметром 0,3 мм, которая не растекается благодаря гидрофобности покрытия на стекле.
Затем игла и кольцо так же быстро переносятся в положение над колодцем с водой и опускаются в него. Одновременно включается насосик, кольцо и игла энергично споласкиваются. Затем они точно так же переносятся в колодец с этанолом, потом в сушильную камеру... Через примерно 5 секунд после начала серии описанных операций игла и кольцо вновь оказываются уже над другой лункой (согласно программе) и все повторяется снова. Легко подсчитать, что для раскапывания всех наших 256-ти проб потребуется не больше 25-ти минут. Интервалы между каплями на стекле равны 0,2 мм. Таким образом, на площади квадрата со стороной 8 мм разместятся все наши олигонуклеотидные «зонды», как их принято называть. (Если бы их было две тысячи, то пришлось бы занять все восемь предметных стекол, а на раскапывание прибор потратил бы немногим более 3-х часов.)
Такая экономия места на стекле (8х8 мм) нужна для того, чтобы все 256 капель поместились в поле зрения бинокулярной лупы (для визуального контроля качества нанесения), а затем перед экраном передающей телевизионной камеры, связанной с компьютером и его монитором.
Описанный прибор (марки GMS-417 Arrayer) появился на рынке сравнительно недавно (в 1998 году) и пока не получил лучшего названия, чем «ЧИП-прибор». Chip в английском языке обозначает стружку, тонкий кусочек. Отсюда и весьма ценимые молодежью картофельные «Чипсы» и «чипы» в микроэлектронике — крошечные пластинки, на которых размещаются тысячи транзисторов. Вообще, разительные успехи технологии электронной промышленности оказали огромное влияние на совершенствование современной научно-исследовательской аппаратуры.
Об этом еще ярче свидетельствует принцип устройства нового прибора того же назначения, что описанный. В нем синтез олигонуклеотидных зондов ведется прямо на стекле под управлением лазерного луча. (Такая возможность была упомянута при изложении идеи химического синтеза олигонуклеотидов.) Утверждается, что с использованием этого метода на площади в 1 квадратный сантиметр можно разместить миллион зондов!
Но вернемся к нашему опыту. Дальнейшая обработка капель на предметном стекле происходит вне ЧИП-прибора. Стекло переносят в камеру для облучения ультрафиолетовым светом. Под его воздействием полимеризуется и «пришивается» к стеклу полиак-риламидный гель. В нем оказываются заполимеризованы и олиго-нуклеотиды. Однако благодаря крупнопористости геля значительная их часть остается доступной для гибридизации. Гель подсушивается.
К молекулам всех иРНК предварительно химически, к концу цепи прикрепляется флюоресцентная метка. Затем стекло покрывают тонким слоем раствора, содержащего смесь всех меченых таким образом иРНК. Выдерживают в условиях, благоприятных для гибридизации. Потом отмывают от всех иРНК, не связавшихся с зондами. Только молекулы нужной иРНК «отыщут» в одной из бывших капель олигонуклеотиды, которые точно соответствуют одному из участков генома, кодировавшего интересующий нас белок. По флюоресценции под действием лазерного луча соответствующую каплю с севшими на нее молекулами иРНК зафиксирует объектив телевизионной камеры. Компьютер укажет их координаты и покажет светящуюся точку на экране монитора.
После чего найденную таким образом иРНК можно освободить из гибрида и использовать для получения кДНК, как это было описано выше. Таким образом задача, обозначенная первой в начале этой главы, решена. Не беда, если количество полученной таким образом кДНК окажется недостаточным для того, чтобы обеспечить весь последующий эксперимент, начинающийся со встраивания в плазмиду. Есть относительно простой способ многократно увеличить это количество по полученному образцу.
Литература
1 Душкин М.П., Иванова М.В. Трансформация перитонических макрофагов в пенистые клетки при внутрибрюшном введении мышам липопротеидов низкой плотности, холестерина и его продуктов окисления. // Патофизиология и эксперимент. терапия. -1993. -N 2. -С.9-11.
2 Дятловская Э.В., Безуглов В.В. Липиды как биоэффекторы. Биохимия. 1998. Т. 67. вып. 1.-С.-3-6.
3 Ершова Л.П., Курбанова Г.Н., Горбунова Н.А. О механизмах посттравматической анемии. // Пат.физиология. 1992. -N 2.-С.-54-55.