Статья: Биосинтез 2Н-меченого бактериородопсина галофильной бактерией Halobacterium halobium
БИОСИНТЕЗ 2Н-МЕЧЕНОГО БАКТЕРИОРОДОПСИНА ГАЛОФИЛЬНОЙ БАКТЕРИЕЙ HALOBACTERIUM HALOBIUM
О. В. МОСИН*
* Московская государственная академия тонкой химической технологии им. М.В. Ломоносова, 117571, Москва, просп. Вернадского, 86;
Осуществлен биосинтез мембранного белка бактериородопсина галофильной бактерией Halobacterium halobium, меченного дейтерием по остаткам [2, 3, 4, 5, 6-2H5]фенилаланина, [3, 5-2H2]тирозина и [2, 4, 5, 6, 7-2H5]триптофана. Комбинацией методов разделения и анализа, включая электрофорез в 12.5% ПААГ с 0.1% ДДС-Na, гель-проникающую хроматографию на сефадексе G-200, обращенно-фазовую ВЭЖХ, спектроскопию 1Н ЯМР и масс-спектрометрию электронного удара метиловых эфиров N-диметиламинонафталин-5-сульфонильных-производных аминокислот, доказаны гомогенность синтезируемого 2Н-меченого БР и селективность включения дейтерия в молекулу.
Ключевые слова: Halobacterium halobium; [2, 3, 4, 5, 6-2H5]фенилаланин, [3, 5-2H2]тирозин, [2, 4, 5, 6, 7-2H5]триптофан, 2Н-меченый бактериородопсин; биосинтез; масс-спектрометрия
ВВЕДЕНИЕ
Ретинальсодержащий белок (хромофор - протонированный альдемин ретиналя с e-аминогруппой Lys-216) - бактериородопсин (БR) выполняет функции АТФ-зависимой транслоказы в клеточной мембране галофильных бактерий Halobacterium halobium [1]. Несмотря на его структурно-функциональную изученность, он остается в центре внимания биотехнологии из-за своей высокой светочувствительности и разрешающей способности и используется в прикладных целях как биологический фотохромный материал [2]. БR также привлекателен, как модельный объект для изучения функциональной активности и структурных свойств мембранных белков в составе искусственно сконструированных энергопреобразующих мембран [3].
Для целого ряда структурно-функциональных исследований с БР целесообразно вводить в молекулу белка изотопную метку дейтерия, позволяющую использовать для последующего анализа изотопного включения метод высокочувствительной масс-спектрометрии электронного удара [4, 5]. Поэтому большое научно-прикладное значение имеет БR, меченный дейтерием по остаткам функционально важных ароматических аминокислот - фенилаланина, тирозина и триптофана, участвующих в гидрофобном взаимодействии полипептидной цепи белка с липидным бислоем клеточной мембраны [6]. 2Н-меченые ароматические аминокислоты могут быть синтезированы с препаративными выходами методом обратного изотопного обмена (1Н-2Н) в молекулах протонированных аминокислот - [2, 3, 4, 5, 6-2Н5]фенилаланин в 85% 2H2SO4 при 500C, [3, 5-2H2]тирозин в 6 н. 2H2SO4 при слабом кипячении реакционной смеси, [2, 4, 5, 6, 7-2H5]триптофан в 75% 2H-меченой трифторуксусной кислоте при 250С [7, 8]. Однако несмотря на изученность современных методов химического синтеза 2Н-меченых ароматических аминокислот, отечественная индустрия индивидуальных 2Н-меченых мембранных белков не получила необходимого развития.
В настоящей работе осуществлен биосинтез БР, меченного дейтерием по остаткам [2, 3, 4, 5, 6-2Н5]фенилаланина, [3, 5-2H2]тирозина и [2, 4, 5, 6, 7-2H5]триптофана для реконструкции искусственных мембран с последующим микропрепаративным выделением, а также исследован уровень дейтерированности молекулы БР методом масс-спектрометрии электронного удара метиловых эфиров N-диметиламинонафталин-5-сульфонильных (Днс)-производных аминокислот с обращенно-фазовой ВЭЖХ.
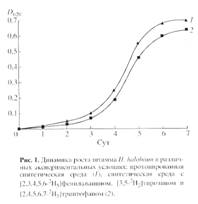
Выбор стратегии биосинтеза 2Н-меченого БР c использованием штамма экстремальной галофильной бактерии Halobacterium halobium определялся целью исследования, связанной с изучением принципиальной возможности получения 2Н-меченых препаратов мембранного белка в микропрепаративном количестве для реконструкции искусственных мембран. При выборе [2, 3, 4, 5, 6-2Н5]фенилаланина, [3, 5-2H2]тирозина и [2, 4, 5, 6, 7-2H5]триптофана в качестве источников дейтерия учитывалась их исключительная важность в гидрофобном взаимодействии молекулы БР с лилипидным бислоем клеточной мембраны, устойчивость к реакциям (1H-2H) обмена в водной среде в условиях выращивания штамма-продуцента, а также возможность применения метода высокочувствительной масс-спектрометрии электронного удара для последующего анализа. В оптимальных условиях выращивания штамма H halobium (синтетическая среда с 4.3 М NaCl, период инкубации 3-4 сут, 35-370С при освещении монохромным светом с 560 нм) в клетке синтезировался каротиноидсодержащий фиолетовый пигмент, по спектральному соотношению белкового и хромофорного фрагментов молекулы D280/D568 1.5:1 идентичный нативному БР. Как показали результаты исследования, рост штамма на синтетической среде (рис. 1, б) ингибировался незначительно по сравнению с контролем (а) на протонированной среде, что существенно упрощает оптимизацию условий биосинтеза 2Н-меченого БР, заключающуюся в эквивалентной замене протонированных ароматических аминокислот среды их дейтерированными аналогами - [2, 3, 4, 5, 6-2Н5]фенилаланином (0.26 г/л), [3, 5-2H2]тирозином (0.2 г/л) и [2, 4, 5, 6, 7-2H5]триптофаном (0.5 г/л).
ОБСУЖДЕНИ Е РЕЗУЛЬТАТОВ
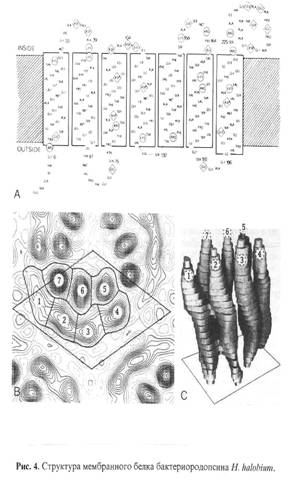
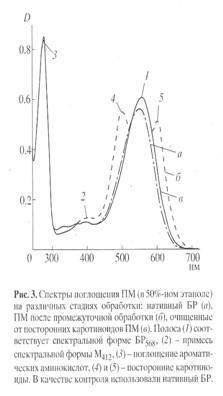
Основными этапами исследования являлись: выращивание штамма экстремальных галофильных бактерий H. halobium на синтетической среде с [2, 3, 4, 5, 6-2Н5]фенилаланином (0.26 г/л), [3, 5-2H2]тирозином (0.2 г/л) и [2, 4, 5, 6, 7-2H5]триптофаном (0.5 г/л), выделение фракции пурпурных мембран (ПМ), отделение от низко- и высокомолекулярных примесей, клеточной РНК, каротиноидов и липидов, фракционирование солюбилизированного в 0.5% ДДС-Na белка метанолом, гель-проникающая хроматография на сефадексе G-200, электрофорез в 12.5% ПААГ с 0.1% ДДС-Na (рис.2). Поскольку белок локализуется в ПМ, освобождение от низкомолекулярных примесей и внутриклеточного содержимого достигали осмотическим шоком клеток дистиллированной водой на холоду после удаления 4.3 М NaCl и последующим разрушением клеточной оболочки ультразвуком при 22 кГц. Последующую обработку клеточного гомогената РНК-азой I (2-3 ед. акт.) проводили для разрушения клеточной РНК. Поскольку фракция ПМ наряду с искомым белком в комплексе с липидами и полисахаридами содержала примесь связанных каротиноидов и посторонних белков, применялись специальные методы фракционирования белка без повреждения его нативной структуры и диссоциации, что существенно усложняло задачу выделения индивидуального БР с применением методов декаротинизации и делипидизации, а также очистки и колоночной хроматографии. Декаротинизация, заключающаяся в многократной обработке ПМ 50% этанолом при -50С, являлась рутинным, но обязательным этапом, несмотря на значительные потери хромопротеина. Использовалось не менее пяти обработок 50% этанолом, чтобы получить спектр поглощения суспензии очищенных от каротиноидов (4) и (5) ПМ (степень хроматографической чистоты 80-85%), показанного на рис. 3 на различных стадиях обработки (б) и (в) относительно нативного БР (а). Образование ретинальпротеинового комплекса в молекуле БР приводит к батохромному сдвигу в спектре поглощения ПМ (рис. 3, в) - основная полоса (1) при максимуме поглощения l 568 нм, вызванная световой изомеризацией хромофора по С13=С14-кратной связи определяется наличием транс-ретинального остатка ретиналя БР568, дополнительная малоинтенсивная полоса (2) при l 412 нм характеризует незначительную примесь образующейся на свету спектральной формы M412 c депротонированной альдиминной связью между остатком транс-ретиналя и белком, а полоса (3) при l 280 нм определяется поглощением ароматических аминокислот в полипептидной цепи белка (для чистого БR соотношение D280/D568 равно 1.5:1).
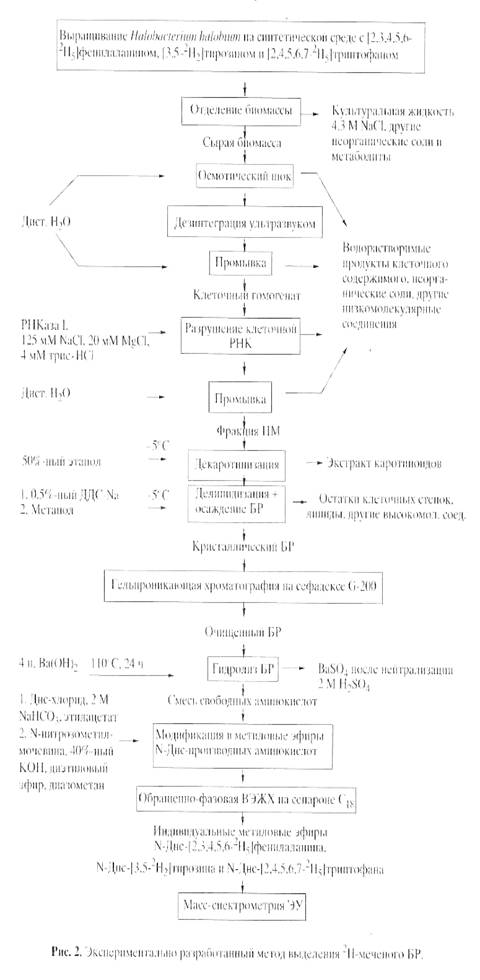
Фракционирование и тщательная хроматографическая очистка белка являлись следующим необходимым этапом. Поскольку БР, будучи трансмембранным белком (Мr 26.7 кД), пронизывает билипидный слой в виде семи a-спиралей, применение сульфата аммония и других традиционных высаливающих агентов не дает положительного результата. Решение проблемы заключалось в переводе белка в растворимую форму солюбилизацией в 0.5% ДДС-Na. Использование ионного детергента ДДС-Na диктовалось необходимостью максимальной солюбилизации белка с комбинированием стадии делипидизации и осаждения в нативном виде, поскольку солюбилизированный в слабоконцентрированном растворе ДДС-Na (0.5%) БР, сохраняет спиральную a-конфигурацию [9]. Поэтому отпала необходимость использования органических растворителей ацетона, метанола и хлороформа для очистки от липидов, а делипидизация и осаждение белка совмещались в одну единственную стадию, существенно упрощающую фракционирование. Значительным преимуществом метода является, что целевой белок в комплексе с молекулами липидов и детергента распределяется в надосадочной жидкости, а другие высокомолекулярные примеси - в непрореагировавшем осадке, легко отделяемом центрифугированием. Фракционирование солюбилизованного в 0.5% ДДС-Na белка с его последующим выделением в кристаллическом виде достигали в три стадии дробным низкотемпературным (-50С) осаждением метанолом, уменьшая концентрацию детергента соответственно в 2.5 и 5 раза. Окончательная стадия очистки БР заключалась в отделении белка от низкомолекулярных примесей методом гель-проникающей хроматографии, для чего БР-содержащие фракции дважды пропускали через колонку с декстрановым сефадексом G-200, уравновешенную 0.09 М Трис-боратным буфером (рН 8.35) с 0.1% ДДС-Na и 2.5 мМ ЭТДА (рис.3). Согласно разработанному методу фракционирования получено 8-10 мг 2Н-меченого БР из 1 г бактериальной биомассы, гомогенность которого удовлетворяла требованиям, предъявляемым для реконструкции мембран и подтверждалась электрофорезом в 12.5% ПААГ с 0.1% ДДС-Na, регенерацией апомембран с транс-ретиналем и обращенно-фазовой ВЭЖХ метиловых эфиров N-Днс-аминокислот. Небольшой выход БР не был препятствием для последующего масс-спектрометрического анализа, однако здесь необходимо подчеркнуть, что для обеспечения высокого выхода белка необходимо наработать большее количество сырьевой биомассы.
Условия проведения гидролиза 2Н-меченого БР определялись необходимостью предотвращения реакций изотопного (1Н-2Н) обмена водорода на дейтерий в молекуле фенилаланина и сохранения остатков триптофана в белке. Рассматривались два альтернативных варианта - кислотный и щелочной гидролиз. Кислотный гидролиз белка в стандартных условиях (6 н. HСl или 8 н. H2SO4, 1100С, 24 ч), как известно, приводит к полному разрушению триптофана и частичному разрушению серина, треонина и некоторых других аминокислот в белке [10], которые для настоящего исследования не играют существенной роли. Модификация этого метода, заключающаяся в добавлении в реакционную среду фенола [11], тиогликолевой кислоты [12], b-меркаптоэтанола [13], позволяет сохранить до 80-85% триптофана. Использование п-толуолсульфокислоты с 0.2% 3-(2-аминоэтил)-индолом или 3 М меркаптоэтансульфокислоты [14] также эффективно для сохранения триптофана (до 93%) [15]. Однако для решения поставленной задачи вышеперечисленные методы непригодны, поскольку обладают существенным недостатком: в условиях кислотного гидролиза с высокой скоростью происходит изотопный обмен ароматических протонов (дейтеронов) в молекулах триптофана, тирозина и гистидина [16], а также протонов при атоме С3 аспарагиновой и С4 глутаминовой кислот [17]. Поэтому даже проведение гидролиза в дейтерированных реагентах (6 н. 2HCl, 4 н. 2H2SO4 в 2H2O) не позволяет получать реальные данные о включении дейтерия в белок.
В условиях щелочного гидролиза (4 н. Ba(OH)2 или 4 н. NaOH, 1100C, 24 ч) реакций изотопного обмена водорода практически не наблюдается (исключением является протон (дейтерон) у атома С2 гистидина, а триптофан не разрушается, что определило выбор метода гидролиза в настоящей работе. Упрощение процедуры выделения смеси свободных аминокислот за счет нейтрализации серной кислотой явилось причиной выбора в качестве гидролизующего агента 4 н. Ba(OH)2. Возможная D,L-рацемизация аминокислот при щелочном гидролизе не влияла на результат последующего масс-спектрометрического исследования уровня дейтерированности молекул аминокислот.
Таблица. Величины пиков (М)+ в масс-спектре электронного удара метиловых эфиров N- Dns-[2, 3, 4, 5, 6-2H5]фенилаланина, N-Dns-[3, 5-2H2]тирозина и N-Dns-[2, 4, 5, 6, 7-2H5]триптофана.
| Соединение |
Величина пика (М)+ |
Интенсив-ность, % | Количество атомов дейтерия | Уровень дейтерированности, % от общего количества атомов водорода |
|
N- Dns-[2, 3, 4, 5, 6-2H5]Phe-Ome |
413 414 415 416 417 418 |
7 18 15 11 14 6 |
1 2 3 4 5 6 |
13 25 38 50 63 75 |
|
N-Dns-[3, 5-2H2]Tyr-OMe |
428 429 430 |
12 15 5 |
- 1 2 |
- 14 29 |
|
N-Dns-[2, 4, 5, 6, 7-2H5]Trp-OMe |
453 454 455 456 457 |
5 6 9 11 5 |
2 3 4 5 6 |
26 38 50 64 77 |
Для изучения уровня дейтерированности 2H-меченого БР использовали метод масс-спектрометрии электронного удара (чувствительность 10-8-10-10 моль анализируемого вещества [18]) после модификации смеси свободных аминокислот гидролизата БР в метиловые эфиры N-Днс-производных аминокислот. Чтобы получить воспроизводимый результат по уровню дейтерированности 2Н-меченого белка, сначала регистрировали полный скан масс-спектр электронного удара смеси метиловых эфиров N-Днс-производных 2Н-меченых аминокислот, по пикам молекулярных ионов которых (М)+ рассчитывали уровень дейтерированности молекулы. Затем проводили разделение метиловых эфиров N-Днс-производных ароматических аминокислот обращенно-фазовой ВЭЖХ и получали масс-спектры электронного удара для каждой индивидуальной аминокислоты. Полный масс-спектр электронного удара смеси метиловых эфиров N-Днс-производных аминокислот, показанный на рис. 4 (сканирование при m/z 50-640, базовый пик m/z 527, 100%), отличался непрерывностью, пики в интервале m/z от 50 до 400 на шкале массовых чисел представлены фрагментами метастабильных ионов, низкомолекулярных примесей, а также продуктами химической модификации аминокислот. Анализируемые 2Н-меченые ароматические аминокислоты, занимающие шкалу массовых чисел m/z от 415 до 456 представлены смесями молекул с различным количеством включенных атомов дейтерия, поэтому молекулярные ионы (М)+ полиморфно расщеплялись на отдельные кластеры со статистическим набором значений m/z зависимости от количества водородных атомов в молекуле. Учитывая эффект изотопного полиморфизма, подсчет уровня дейтерированности молекул аминокислот проводили по наиболее распространенному пику молекулярного иона (М)+ в каждом кластере с математически усредненной величиной (М)+ (рис. 4) - для фенилаланина пик молекулярного иона определялся (М)+ при m/z 417, 14% (вместо (М)+ при m/z 412, 20% для немеченого производного (пики немеченых аминокислот не показаны)), тирозина - (М)+ при m/z 429, 15% (вместо (М)+ при m/z 428, 13%), триптофана - (М)+ при m/z 456, 11% (вместо (М)+ при m/z 451, 17%). Уровень дейтерированности, соответствующий увеличению молекулярной массы составил для тирозина два, фенилаланина и триптофана - пять атомов дейтерия. Полученные данные по уровню дейтерированности фенилаланина, тирозина и триптофана позволяют сделать вывод о высокой селективности включения 2H-меченых ароматических аминокислот в молекулу БР: дейтерий детектировался во всех остатках ароматических аминокислот (таблица). Обсуждая полученные результаты, необходимо подчеркнуть, что присутствие в масс-спектре пиков (M)+ протонированных и полудейтерированных аналогов фенилаланина с (M)+ при m/z 413-418, тирозина с (M)+ при m/z 428-430 и триптофана с (M)+ 453-457 с различными вкладами в уровни дейтерированности молекул, свидетельстствует о сохранении небольшой доли минорных путей биосинтеза de novo, приводящим к разбавлению дейтериевой метки и, по-видимому, определяется самими условиями биосинтеза 2Н-меченного БР (таблица).
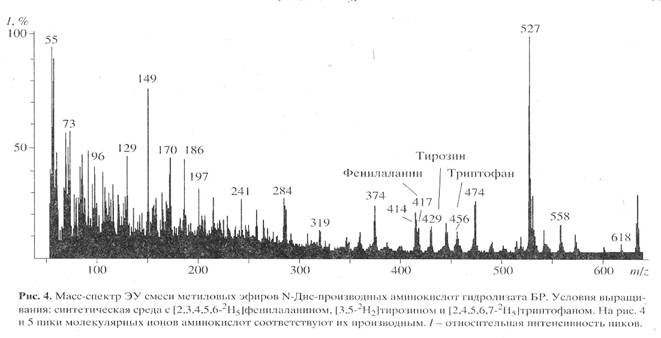
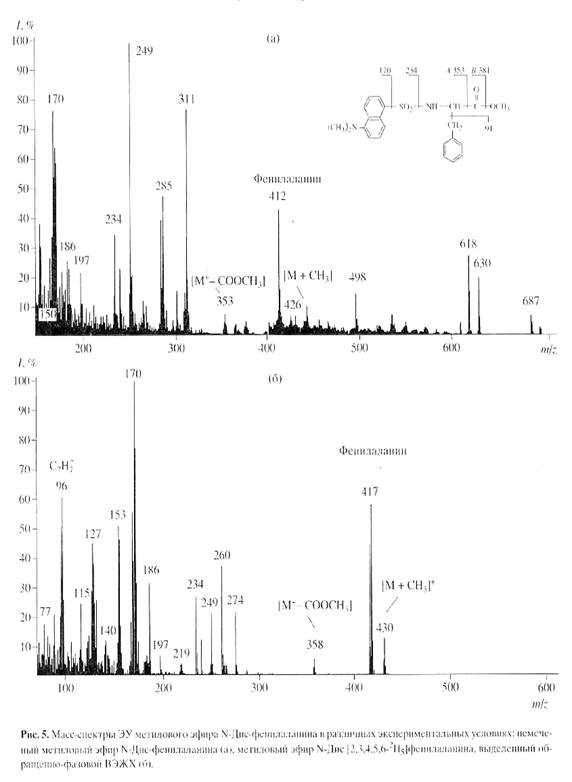
Согласно данным масс-спектрометрического анализа, пики молекулярных ионов (М)+ метиловых эфиров N-Днс-производных ароматических аминокислот обладали очень низкой интенсивностью и полиморфно расщеплялись, поэтому области их молекулярного обогащения были сильно уширены. Кроме этого, масс-спектры компонентов смеси аддитивны, поэтому смеси можно анализировать, только если имеются спектры различных компонентов, записанные в тех же условиях [8]. Проводимые вычисления предусматривают решение системы из n уравнений с n неизвестными для смеси из n компонентов. Для компонентов, концентрация которых превышает 10 мол.%, правильность и воспроизводимость результатов анализа составляет +0.5 мол.% (при доверительной вероятности 90%). Поэтому для получения воспроизводимого результата необходимо хроматографически выделять индивидуальные производные 2Н-меченых аминокислот из белкового гидролизата. Для решения поставленной задачи использовали метод обращенно-фазовой ВЭЖХ на октадецилсилановом селикагеле силасорб С18, эффективность которого подтверждалась разделением смеси метиловых эфиров N-Днс-производных 2Н-меченых аминокислот из других микробных объектов, как метилотрофные бактерии и микроводоросли [19]. Метод удалось адаптировать к условиям хроматографического разделения смеси метиловых эфиров N-Днс-производных аминокислот гидролизата БР, заключающийся в оптимизации соотношения элюентов, форме градиента и скорости элюции с колонки. Наилучшее разделение достигалось при градиентной элюции метиловых эфиров N-Dns-производных аминокислот смесью растворителей ацетонитрил : трифторуксусная кислота = 100 : 0.1 - 0.5, об.%. При этом удалось разделить триптофан и трудно разрешимую пару фенилаланин/тирозин. Степени хроматографической чистоты выделенных метиловых эфиров N-Днс-[2, 3, 4, 5, 6-2H5]фенилаланина, N-Днс-[3, 5-2H2]тирозина и N-Днс-[2, 4, 5, 6, 7-2H5]триптофана составили 89, 91 и 90% при выходах 78-85%. Полученный результат подтвердил рис. 4, б на котором приведен масс-спектр электронного удара метилового эфира N-Днс-[2, 3, 4, 5, 6-2H5]фенилаланина, выделенного обращенно-фазовой ВЭЖХ (сканирование при m/z 70-600, базовый пик m/z 170, 100%) (масс-спектр приведен относительно немеченого метилового эфира N-Днс-фенилаланина (а), сканирование при m/z 150-700, базовый пик m/z 250, 100%). Доказательством включения дейтерия в молекулу фенилаланина является пик тяжелого молекулярного иона метилового эфира N-Днс-фенилаланина ((М)+ при m/z 417, 59% вместо (М)+ при m/z 412, 44% для немеченого производного фенилаланина) и дополнительный пик бензильного фрагмента фенилаланина С7Н7+ при m/z 96, 61% (вместо m/z 91, 55% в контроле (не показан)) (рис. 5, б). Пики второстепенных фрагментов различной интенсивности со значениями m/z 249, 234 и 170 принадлежат к продуктам вторичного распада дансильного остатка до N-диметиламинонафталина, низкоинтенсивный пик (M - COOCH3)+ при m/z 358, 7% (m/z 353, 10%, контроль) является продуктом отщепления карбоксиметильной СООСН3-группы из метилового эфира N-Днс-фенилаланина, а пик (M + CH3)+ при m/z 430, 15% (m/z 426, 8%, контроль) - продуктом дополнительного метилирования по a-аминогруппе фенилаланина (рис. 5, б). Согласно данным масс-спектра, разница между молекулярной массой легкого и тяжелого пиков [M]+ метилового эфира N-Днс-фенилаланина составляет пять единиц, что совпадает с полученными ранее данными по уровню дейтерированности исходного [2, 3, 4, 5, 6-2H5]фенилаланина, добавляемого в среду выращивания (масс-спектрометрические данные по уровням дейтерированности [2, 3, 4, 5, 6-2H5]фенилаланина, [3, 5-2H2]тирозина и [2, 4, 5, 6, 7-2H5]триптофана подтверждены спектроскопией 1Н ЯМР и находятся в корреляции).
Полученные экспериментальные данные, свидетельствуют о высокой эффективности включения дейтерия в молекулу БР. Планируется использовать полученные дейтерированные препараты БР для реконструкции в 2Н2О функционально активных систем мембранных белков с очищенными 2Н-мечеными жирными кислотами и другими биологически активными соединениями.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объектом исследования служил каротиноидсодержащий штамм экстремальных галофильных бактерий Halobacterium halobium ЕТ 1001, полученный Яном Раапом (Лейденский университет, Голландия). Штамм модифицирован селекцией отдельных колоний на твердой (2% агар) пептоновой среде с 4.3 М NaCl [20].
В работе использовали D,L-аминокислоты (Reanal, Венгрия), АМФ и УМФ (Sigma, США). Для синтеза производных аминокислот использовали N-диметиламинонафталин-5-сульфохлорид (Днс-хлорид) (Sigma, США) и диазометан, получаемый из N-нитрозометилмочевины (Merck, ФРГ). L-[2, 3, 4, 5, 6-2H5]фенилаланин (90 ат.% 2Н), L-[3, 5-H2]тирозин (96 ат.% 2Н) и L-[2, 4, 5, 6, 7-2H5]триптофан (98 ат.% 2Н) (способы получения указаны в работах [21, 22]) предоставлены к. х. н. А. Б. Пшеничниковой (МГАТХТ). Масс-спектры метиловых эфиров N-Днс-производных аминокислот получали методом электронного удара на приборе Hitachi MB-80 A (Япония) при энергии ионизирующих электронов 70 эВ, ускоряющем напряжении 8 кВ и температуре катодного источника 180-2000С. Сканирование анализируемых образцов проводили при разрешении 7500 усл. ед., используя 10%-ную резкость изображения. Спектры 1Н-ЯМР регистрировали в 2Н2О на приборе Bruckman WM-250 (ФРГ) с рабочей частотой 70 МГц, химические сдвиги протонов (d) приведены в миллионных долях по отношению к Ме4Si. УФ-спектры регистрировали на спектрофотометре Beckman DU-6 (США) в диапазоне длин волн 200-750 нм. Центрифугирование осуществляли на центрифуге Т-24 (Германия) с охлаждением при -40С. Обращенно-фазовую ВЭЖХ проводили на жидкостном хроматографе Knauer (ФРГ), снабженным насосом Knauer, УФ-детектором UF-2563 и интегратором Shimadzy СR-3A (Япония), используя колонку 250 x 10 мм с неподвижной обращенной фазой сепарон С18 (Kova, Чехоcловакия); элюент: (А) - ацетонитрил : трифторуксусная кислота = 100 : 0.1 - 0.5, об.% и (В) - ацетонитрил = 100 об.%; скорость элюции - 1.5 мл/мин: от 0 до 20% В - 5 мин, от 20 до 100% В - 30 мин, 100% В - 5 мин, от 100 до 0% В - 2 мин, 0% В - 10 мин. ТСХ проводили на хроматографических пластинках с закрепленным слоем флуоресцентного носителя Silufol UV-254 (Kavalier, Чехословакия) в системе (Г): н-бутанол : уксусная кислота : вода = 12 : 3 : 5, об.%. Электрофорез проводили в 12.5% ПААГ с 0.1% ДДС-Na в соответствие с протоколом фирмы LKB (Швеция). Количественное определение содержания белка выполняли сканированием прокрашенного в растворе кумасси-голубой R-250 электрофоретического геля на лазерном денситометре Beckman CDS-200 (США). Бактериальный рост изучали по оптической плотности бактериальной суспензии, измеренной при 540 нм на спектрофотометре Beckman DU-6 (США). Процедура выделения БР проводилась с использованием светозащитной лампы, снабженной оранжевым светофильтром ОРЖ -1X (200 x 0.5 мм).
2Н-Меченый БР (выход 8-10 мг с 1 грамма бактериальной биомассы) получали на синтетической среде, заменяя протонированные фенилаланин, тирозин и триптофан их дейтерированными аналогами - L-[2, 3, 4, 5, 6-2H5]фенилаланином, L-[3, 5-2H2]тирозином и L-[2, 4, 5, 6, 7-2H5]триптофаном (г/л): D,L-аланин - 0.43, L-аргинин - 0.4, D,L-аспарагиновая кислота - 0.45, L-цистеин - 0.05, L-глутаминовая кислота - 1.3, L-глицин - 0.06, D,L-гистидин - 0.3, DL-изолейцин - 0.44, L-лейцин - 0.8; L-лизин - 0.85, D,L-метионин - 0.37, DL-фенилаланин 0.26, L-пролин 0.05, D,L-серин 0.61, D,L-треонин 0.5, L-тирозин 0.2, D,L-триптофан 0.5, D,L-валин - 1.0; АМФ - 0.1, УМФ - 0.1, NaCl - 250, MgSO4 . 7H2O - 20, KСl - 2, NH4Cl - 0.5, KNO3 - 0.1, KH2PO4 - 0.05, K2HPO4 - 0.05, Na+-цитрат - 0.5, MnSO4 . 2H2O - 3 x 10-4, CaCl2 . 6H2O - 0.065, ZnSO4 . 7H2O - 4 x 10-5, FeSO4 . 7H2O - 5 x 10-4, CuSO4 . 5H2O - 5 x 10-5, глицерин - 1.0; биотин - 1 x 10-4, фолиевая кислота - 1.5 x 10-4, витамин В12 - 2 x 10-5. Среду автоклавировали 30 мин при 0.5 ати, рН доводили 0.5 М КОН до 6.5-6.7. Синтез проводили в колбах Эрленмейера, вместимостью 500 мл (объем реакционной смеси 100 мл) 3-4 сут при 35-370С в условиях интенсивной аэрации на орбитальном шейкере Biorad 380-S (Венгрия) и освещении монохромными лампами ЛДС-40 (3 x 1.5 лк).
Выделение фракции пурпурных мембран (ПМ). Биомассу (1 г) промывали дистиллированной водой и осаждали центрифугированием (1500 g, 20 мин). Осадок суспендировали в 100 мл дистиллированной воды и выдерживали 1 сут при 40С. Реакционную смесь центрифугировали (1500 g, 15 мин), осадок ресуспендировали в 20 мл дистиллированной воды и дезинтегрировали ультразвуком (22 кГц, 3 x 5 мин) в водяной бане со льдом (00С). Клеточный гомогенат после промывки дистиллированной водой суспендировали в 10 мл буфера 125 мМ NaCl, 20 мМ MgCl2, 4 мМ Трис-HCl, (рН 8.0), добавляли 5 мкг РНК-азы I (2-3 ед. акт.) и инкубировали 2 ч при 370С. Затем добавляли 10 мл того же буфера, выдерживали 14-16 ч при 40С. Водную фракцию отделяли центрифугированием (1500 g, 20 мин), осадок ПМ обрабатывали 50% этанолом (5 x 7 мл) при -50С с последующим отделением растворителя. Процедуру повторяли трижды до получения бесцветных промывных вод. Содержание белка определяли спектрофотометрически по соотношению D280/D568 (e280 1.1 x 105 и e568 6.3 x 104 M-1 см-1 [23]). Регенерацию ПМ проводили как описано в работе [24]. Выход фракции ПМ 120 мг (х. ч. 80-85%).
БР выделяли по методу Остерхельта [25], модифицированного нами [24], солюбилизируя фракцию ПМ (в Н2О) (1 мг/мл) в 1 мл 0.5% ДДС-Na, смесь инкубировали 7-9 ч при 370С с последующим центрифугированием (1200 g, 15 мин). Осадок отделяли, к супернатанту добавляли дробными порциями метанол (3 x 100 мкл) при 00С, выдерживали 14-15 ч при -50С и центрифугировали при охлаждении (1200 g, 15 мин). Фракционирование проводили трижды, уменьшая концентрацию 0.5% ДДС-Na до 0.2 и 0.1%. Кристаллический белок (8-10 мг) промывали холодной дистиллированной водой (2 x 1 мл) и центрифугировали (1200 g, 15 мин).
Очистку БР осуществляли методом гель-проникающей хроматографии на откалиброванной колонке с габаритными размерами 150 x 10 мм; неподвижная фаза - Сефадекс G-200 (Pharmaсia, США) (удельный объем упакованных гранул - 30-40 ед на 1 г сух. сефадекса) с ручным отбором проб. Колонку уравновешивали буферным раствором, содержащим 0.1% ДДС-Na и 2.5 мМ ЭТДА. Пробу белка растворяли в 100 мкл буферного раствора и элюировали 0.09 М Трис-боратным буфером, содержащим 0.5 М NaCl с pH 8.35 (I = 0.075) со скоростью 10 мл/см2 x ч. Объединенные белковые фракции подвергали лиофильной сушке, запаивали в стеклянные ампулы (10 x 50 мм) и хранили в темноте при -40С.
Гидролиз БР. 4 мг белка помещали в стеклянные ампулы размером 10 x 50 мм, добавляли 5 мл 4 н. Ba(OH)2 и выдерживали 24 ч при 1100С. Реакционную смесь суспендировали в 5 мл горячей дистиллированной воды и нейтрализовали 2 н. H2SO4 до рН 7.0. Выпавший осадок сульфата бария отделяли центрифугированием (200 g, 10 мин), супернатант удаляли при 10 мм рт. ст.
N-Днс-производные аминокислот. К 4 мг сухого гидролизата БR в 1 мл 2 M NaHCO3 (рН 9-10) порциями при перемешивании добавляли 25.6 мг Днс-хлорида в 2 мл ацетона. Реакционную смесь выдерживали 1 ч при перемешивании при 400С, подкисляли 2 н. HCl до рН 3 и экстрагировали этилацетатом (3 x 5 мл). Объединенный экстракт промывали дистиллированной водой до рН 7.0 (2 x 1 мл), сушили безводным сульфатом натрия, растворитель удаляли при 10 мм. рт. ст. Выход 15.3 мг (78%).
Метиловые эфиры N-Днс-производных аминокислот. Для получения диазометана к 20 мл 40% КОН в 40 мл диэтилового эфира добавляли 3 г влажной N-нитрозометилмочевины и перемешивали 15-20 мин на водяной бане со льдом. После окончания газовыделения эфирный слой отделяли, промывали дистиллированной водой до рН 7.0, сушили безводным сульфатом натрия и использовали для обработки N-Днс-производных аминокислот. Выход 17.4 мг (69%).
L-[2, 3, 4, 5, 6-2H5]фенилаланин. 40 г фенилаланина растворяли в 300 мл 85% 2Н2SО4 (в 2Н2О) и нагревали с обратным водяным холодильником при 50-600С при перемешивании 3 сут. По окончании реакционную смесь охлаждали, нейтрализовали 30% NH4OН до рН 5.5. Продукт экстрагировали этанолом. Выход 24 г (58.7%). Т пл. 271-273, [a]d25 4.47 (с 1-2, 5 М НСl). рKa 2.20 (СООН), 9.31 (NH2). Rf 0.6 (Г). УФ-спектр (0.1 М НCl): lmax нм (e М-1 см-1): 257.5 (e 195). 1Н-ЯМР: d 3.25 (2H, Hb), 4.4 (1H, Ha), 7.2-7.4 (0.07Н), УД 90 ат.% 2Н. Масс-спектр (M)+ m/z (I, %): 165 (34), метиловый эфир N-Dns-[2, 3, 4, 5, 6-2H5]фенилаланина: 417 (14), 418 (6).
L-[3, 5-2H2]тирозин. 100 г тирозина растворяли в 150 мл 3 М 2Н2SO4. Реакционную смесь нагревали 2 сут при 40-500С с обратным водяным холодильником в токе азота. По окончании нейтрализовали 28% NH4OH до рН 4.5 и охлаждали 1 сут при 40С. Кристаллический продукт фильтровали, промывали 2Н2О и сушили при 10 мм рт ст. Выход 90 г (86.5%). Т пл. 316 - 317, [a]d25 10.0 (с 2, 5 М НСl). рKa 2.20 (СООН), 9.21 (NH2). Rf 0.45 (Г). УФ-спектр (0.1 М Нcl) lmax нм (e М-1 см-1): 223 (e 8200) и 274.5 (e 1340). 1Н-ЯМР: d 3.32 (2H), d 4.35 (1H), d 6.9 (1H), d 7.2 (2H), УД 96 ат.% 2Н. Масс-спектр (M)+ m/z (I, %): 181 (21), метиловый эфир N-Dns-[3, 5-H2]тирозина: 429 (15), 430 (5).
L-[2, 4, 5, 6, 7-2H5]триптофан. К 40 мл 100% 2Н2О добавляли при 40С и перемешивании 80 мл трифторуксусного ангидрида (0.5 моль) и выдерживали 2 ч при 40С, затем дробными порциями добавляли 25 г триптофана. Реакционную смесь выдерживали 3 сут в темноте при 220С, расстворитель удаляли при 10 мм рт., нейтрализовали 30% NH4OH до 5.9, охлаждали 1 сут при 40С. Кристаллический продукт фильтровали, промывали 2Н2О и сушили при 10 мм рт. ст. Выход 19 г (60.3%). Т пл. 283-285, [a]d25 2.8 (с 1-2, 1 М НСl). рKa 2.46 (СООН), 9.41 (NH2). Rf 0.5 (Г). УФ-спектр (0.1 М НCl) lmax нм (e М-1 см-1): 218 (e 33500), 278 нм (e 5550), 287.5 (e 4550). 1Н-ЯМР: d 3.4 (2H, Hb), 4.4 (1H, Ha), 7.3 (1H, He), 7.2-7.4 (0.1Н, In-Н), УД 98 ат.% 2Н. Масс-спектр (M)+ m/z (I, %): 204 (28), метиловый эфир N-Dns-[2, 4, 5, 6, 7-2H5]триптофана: 455 (9), 456 (11).
СПИСОК ЛИТЕРАТУРЫ
1. Oesterhelt D., Stoeckenius W. // Nature. 1971. V. 233. ¹ 89. P.149-160
2. Spudich J. L. // Ann. Rev. Biophys. Chem. 1988. V. 17. ¹ 12. P.193-215.
3. Karnaukhova E.N., Niessen W. M.A., Tjaden U.R. // Anal. Biochem. 1989. V. 181. ¹ 3. P. 271-275
4. Мосин О.В., Складнев Д.А., Егорова Т.А., Швец В.И. // Биоорган. химия. 1996. Т. 22. ¹ 10-11. С. 856-869.
5. Mosin O.V., Karnaukhova E.N., Pshenichnikova A.B., Reshetova O.S. Electron impact mass-spectrometry in bioanalysis of stable isotope labeled bacteriorhodopsin / in: 6th Intern. Conf. on Retinal proteins. 1994. Leiden, the Netherlands, P. 115.
6. Hardy J. P., Knight A.E.W., Ghiggino K.P., Smith T.A., Rogers P.J. // Photochem. Photobiol. 1984. V. 39. ¹ 1. P. 81-88.
7. Rosenbach V., Goldberg R., Gilon C., Ottolenghi M. // Photochem. Photobiol. 1982. V. 36. ¹ 6. P. 197-201.
8. Мосин О.В., Складнев Д.А., Егорова Т.А., Швец В.И. // Биотехнология. 1996. № 10. С. 24-40.
9. Первушин К.В., Арсеньев А.С. // Биоорган. химия. 1995. Т. 21. № 10. С. 83-111.
10. Звонкова Е.Н., Зотчик Н.В., Филлипович Е.И., Митрофанова Т.К., Мягкова Г.И., Серебренникова Г.А. Химия биологически активных природных соединений. М.: Химия, 1970. С. 65-68.
11. Muramoto K., Sunahara S., Kamiya H. // Agric. Biol. Chem. 1987. V. 51. ¹ 6. P. 1607-1616.
12. Matsubara H., Sasaki R.M. // Biochim. Biophys. Res. Com. 1969. V. 35. ¹ 10. P. 175-177.
13. Ng L.T., Pascaud A., Pascaud M. // Anal. Biochem. 1987. V. 167. ¹ 2. P. 47-52.
14. Liu T.Y., Chang Y.H. // J. Biol. Chem. 1971. V. 246. ¹ 2. P. 2842-2848.
15. Simpson R.J., Neuberger M.R., Liu T.Y. // J. Biol. Chem. 1976. V. 251. ¹ 3. P. 1936-1938.
16. Пшеничникова А.Б., Карнаухова Е.Н., Звонкова Е.Н., Швец В.И. // Биоорганическая химия. 1995. Т. 21. ¹ 3. С. 163-178.
17. Cohen J.S., Putter I. // Biochim. Biophys. Acta. 1970. V. 222. ¹ 1. P. 515-520.
18. Mosin O.V., Skladnev D.A., Shvets V.I. // Biosc. biotechnol. biochem. 1998. V. 62. № 2. P. 225-229.
19. Егорова Т.А., Мосин О.В., Еремин С.В., Карнаухова Е.Н., Звонкова Е.Н., Швец В.И. // Биотехнология. 1993. № 8. С. 21-25.
20. Мосин О.В., Складнев Д.А., Швец В.И. // Приклад. биохим. микробиол. 1999. Т. 35. ¹ 1. С. 34-42.
21. Griffiths D.V., Feeney J., Roberts G.C., Burgen A.S. // Biochim. Biophys. Acta. 1976. V. 446. ¹ 4. P. 479-585.
22. Matthews H.R., Matthews K.S, Opella S.J. // Biochim. Biophys. Acta. 1977. V. 497. ¹ 23. P. 1-13.
23. Tokunada F., Ebrey T. // Biochemistry. 1978. V. 17. ¹ 10. P. 1915-1922.
24. Мосин О.В., Егорова Т.А., Чеботаев Д.В., Складнев Д.А., Юркевич А.М., Швец В.И. // Биотехнология. 1996. ¹ 4. С. 27-35.
25. Oesterhelt D., Hess B. // Eur. J. Biochem. 1973. V. 37. ¹ 1. P. 316-326
BIOSYNTHESIS OF 2H-LABELED BACTERIORHODOPSIN BY HALOPHILIC BACTERIUM Halobacterium halobium
O. V. МOSIN
* Moscow State Academy of Fine Chemical Technology named after M.V. Lomonosov, 117571, Moscow, Vernadskogo prosp., 86;
The biosynthesis of 2H-labeled membrain protein bacteriorhodopsin by halophilic bacterium Halobacterium halobium, labeled with deuterium on residues of [2, 3, 4, 5, 6-2H5]phenylalanine, [3, 5-2H2]tyrosine, and [2, 4, 5, 6, 7-2H5]tryptophan was carried out. The combination of preparative and analitic methods including elecrtophoresis in 12.5% PAAG with 0.1% SDS-Na, gel filtration chromatography on Sephadex G-200, reverse-phase HCLP, 1Н NMR spectroscopy, and electron impact mass-spectrometry of methyl esters of N-Dns-derivatives of amino acids was used to prove the homogenity of the synthesized product, and the selectivity of the introduction of deuterium into the molecule.