Реферат: Краткая история развития катализа и теорий, объясняющих это явление
Краткая история развития катализа и теорий, объясняющих это явление.
Краткая история развития катализа и теорий, объясняющих это явление
Катализ – универсальное и очень разнообразное явление, широко распространенное в природе и используемое человечеством за тысячи лет до осознания сути каталитических процессов. Наилучшим примером служит ферментативный катализ. Люди используют биологические катализаторы – ферменты - тысячи лет в процессах брожения (для приготовления, например, молочно-кислых продуктов).
Что же такое катализ и катализатор?
В литературе встречаются различные определения катализа и катализатора. Приведем некоторые из них.
В. Оствальд: Катализатор – это такое соединение, которое ускоряет химическую реакцию, не влияя на положение равновесия.
П. Сабатье: Катализатор – вещество или система, которая изменяет скорость реакции, участвуя в последовательности стадий, но не превращаясь в продукты.
Г.К. Боресков: Феноменологически катализ – это возбуждение химических реакций или изменение их скорости под влиянием веществ – катализаторов, многократно вступающих в промежуточное химическое взаимодействие с участниками реакции и восстанавливающих после каждого цикла промежуточных взаимодействий свой состав.
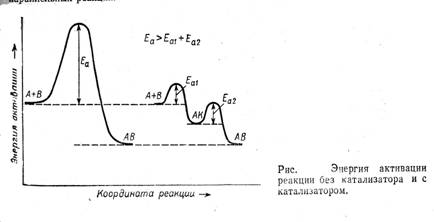
Последнее определение включает два существенных момента: катализатор входит в состав промежуточных соединений, но не фигурирует в стехиометрическом уравнении основной реакции и, следовательно, не влияет на равновесие основной реакции и не расходуется в ней. Катализатор за счет участия в образовании интермедиатов обеспечивает протекание реакции по другому пути, имеющему более низкую наблюдаемую энергию активации, и значит более высокую скорость превращения реагентов в продукты (см. рис. 1).
Химический (небиологический) катализ принято делить на гомогенный и гетерогенный. Гомогенный катализ – это такой процесс, протекающий в одной фазе, в которой находятся реагенты и катализатор. Гетерогенный катализ – это процесс, в котором катализатор – твердое вещество, а реагенты могут быть жидкими или газообразными. С участием гомогенных катализаторов протекают гомогенно-каталитические процессы, а с участием гетерогенных – гетерогенно-каталитические. Эту классификацию нельзя путать с классификацией реакционных систем по фазности (гомофазные – одна фаза, гетерофазные – несколько фаз и есть границы раздела фаз). Системы, в которых протекают гомогенно-каталитические процессы могут быть как гомофазными, так и гетерофазными.
Слово «катализ», вероятно, впервые введено в 16 веке химиком А. Либавиусом в его учебнике «Алхимия» и имело значение «разложение» или «разрушение». В 1835 г. этот термин узаконен И. Берцелиусом для реакций, протекающих в присутствии посторонних соединений, которые сами как будто в реакции не участвуют. Точнее Берцелиус писал о каталитической силе, приводящей к разложению тел. Примерно в то же время Митчерлих ввел термин «контактное действие».
На самом деле даже “небиологический”, т.е. неферментативный катализ был известен задолго до Либавиуса и тем более Берцелиуса.
Первый известный нам пример небиологического каталитического процесса – синтез диэтилового эфира из спирта при участии серной кислоты (VIII в., Джабир ибн Хайам).
C2H5OH + HO-C2H5 → H2O + C2H5-O-C2H5
Вторично эта реакция была открыта в 1540 г. Валерием Кордусом и получила технологическое оформление в работах С. Фробениуса.
В XVII и XVIII вв. во время создания научных основ химии было открыто несколько каталитических реакций с участием небиологических катализаторов. Так, в 1666 г. А. Лефебр и Н. Лемери разработали камерный способ синтеза серной кислоты базируясь на предыдущих разработках. Это XVII век. И только в конце следующего, XVIII века, механизм синтеза серной кислоты изучен М. Клеманом и Х. Дезормом. Они доказали, что в реакции участвуют не только реагенты (SO2 и O2 ), но и оксиды азота:
NO2 + SO2 + H2O → NO + H2SO4
NO + 1/2O2 → NO2
Клеман и Дезорм указали, что оксиды азота – “только орудие для полного окисления серной кислоты”, и отметили два важных принципа катализа: нестехиометричность и цикличность действия оксидов азота.
Механизм вышеупомянутой реакции дегидратации этилового спирта в диэтиловый эфир изучал А. Геннель в лаб. М. Фарадея в 1828 г.
Краткая история открытия каталитических реакций представлена в таблице1.
Таблица 1.
Краткая история катализа
| Год | Авторы | Процесс / катализатор |
| 1 | 2 | 3 |
| Гомогенный катализ | ||
| 1746 | Дж. Робек |
SO2 + 0,5 O2 → SO3 / NO2 |
| 1782 | К. Шееле |
RCOOH + R’OH → RCOOR’ + H2O / Минеральн. к-ты |
| 1878 | А.М. Бутлеров |
m CnH2n → -(CnH2n)-m / H2SO4 |
| 1881 | М.Г. Кучеров |
С2Н2 + Н2О → СН3СНО / Hg2+ |
| 1928-1929 | Ю. Ньюленд |
2 С2Н2 → СН2=СНС≡СН / Cu(I) |
| 1938 | О. Роелен |
CnH2n + СО + Н2 → CnH2n+1СНО / Со2(СО)8 |
| 1939-1945 | В. Реппе |
С2Н2 + СО + НХ→ СН2=СНСОХ / Ni(CO)4 С2Н4 + СО + НX → СН3СН2СОХ / Ni(0), Co(0) (X – OH, OR, NR2, SR) 4 С2Н2 → циклооктатетраен / Ni(CN)2 С2Н2 + 2 CH2O → HOCH2C≡CCH2OH / Cu2C2 |
| 1953-1955 | К. Циглер, Д. Натта |
m α-CnH2n → -(CnH2n)-m / TiCl3-AlR3, TiCl4-AlR3, гомог. и гетерог. катализаторы полим-ции 1-алкенов и диенов |
| 1959 | Ю. Смидт, И.И. Моисеев |
С2Н4 + 0,5 О2 → СН3СНО / PdCl2-CuCl2 |
| 1960 | И.И. Моисеев, |
С2Н4 + CH3COOH + 0,5 О2 → СН2=СНОOCCH3 + H2O / PdCl2-CuCl2-CH3COONa |
| 1960 | Фирма БАСФ АГ |
CH3OH + CO → СН3СОOH / CoI2 |
| 1970 |
Ф.Е. Паулик, Д.Ф. Роз, фирма «Монсанто» |
CH3OH + CO → СН3СОOH / Rh(I)-CH3I |
| 1972 | Фирма «Халкон» | Сопряж. процесс получ. стирола и оксида пропилена |
| Гетерогенный катализ | ||
| 1778 | Ж. Пристли |
СnH2n+1OH → СnH2n + H2O / глина (алюмосиликат) |
| 1796 | М. Ван-Марум |
С2Н5ОН → СН3СНО + Н2 / металлы: Ag,Cu, Ce, Fe, Ni, Pb, Sn, Mn |
| 1831 | П. Филлипс |
SO2 + 0,5 O2 → SO3 / Pt |
| 1844 | М. Фарадей |
С2Н4 + Н2 → С2Н6 / Pt |
| 1863 | Г. Дебус |
HCN + Н2 → СH3NН2 / Pt |
| 1867 | Г. Дикон |
4 HCl + O2 → 2 Сl2 + 2 H2O / Cu |
| 1867 | А. Гофман |
CH3OH + 0,5 O2 → СH2O + H2O / Pt |
| 1877 | М.М. Зайцев |
Гидрирование органических соед-й Н2 в жидк. фазе/ Pd, Pt |
| 1890 | ? |
СН3ОН+ 0,5 O2 → СH2O + H2O ( Н2) / металлы: Ag,Cu |
| 1900-е | П. Сабатье | Гидрирование и окисление орг.соед-й / Ni |
| 1903 | В. Оствальд |
2 NH3 + 3 O2 → NO + NO2 + 3 H2O / Pt |
| 1908-1914 | Маттиаш, Габер, Бош (БАСФ) |
3 H2 + N2 = 2 NH3 / FeO+Al2O3+Ca+K+SiO2 |
| 1923 | Бош (БАСФ) |
СО + 2 Н2 → СH3OН / ZnO∙Cr2O3, ZnO∙CuO∙Cr2O3 |
| 1930 | Э. Фишер, Г. Тропш |
n СО + (2n+1) Н2 → СnH2n+2 + n Н2O / Co 2n СО + (n+1) Н2 → СnH2n+2 + n CO2 / Fe продукты содержат алкены и кислородсодерж. соед-я |
| 1931 | Т.Е. Лефорт |
С2Н4 + 0,5 О2 → (СН2СН2)О / Ag/ носитель |
| 1 | 2 | 3 |
| 1930-е |
Катал. крекинг, риформинг / MoO3∙Al2O3, Pt/ Al2O3 |
|
| 1940е | Катал. Гидрокрекинг | |
| 1955 | Фирма «Халкон» |
С6Н6 + 4,5 О2 → Малеинов. анг-д + 2 Н2О + 2 СО2 / V2O5 + промоторы |
| 1960 | Фирма «Сохио» |
C3H6 + 1,5 О2 + NH3 → CH2=CHCN + 3 Н2О + 2 СО2 / MoO3∙Bi2O3∙P2O5 + добавки |
| 1967 | Фирма «Баер» |
С2Н4 + CH3COOH + 0,5 О2 → СН2=СНОOCCH3 + H2O / Pd(OAc)2-CH3COOK/ SiO2 |
Разделение процессов по принципу –
- гомогенный катализ
- гетерогенный катализ – сложилось исторически.
Действительно, на первый взгляд на ранних этапах развития катализа между этими процессами было мало общего.
Так, в гомогенном катализе реакции протекают в основном в растворе, превращение субстрата на металле-катализаторе аналогичны превращению лиганда в координационной сфере металла (то, что наблюдается в координационной химии). И тут можно получить информацию об интермедиатах процесса, о механизме элементарного акта.
В гетерогенном катализе – при поверхностном рассмотрении – нет ничего общего. Здесь основной стадией является адсорбция субстрата на активном центре и некоторую информацию получают из кинетических данных.
Такое разделение заставило исследователей рассматривать отдельно гомогенный катализ и гетерогенный катализ и не способствовало поиску общих закономерностей.
На самом деле здесь много общего. И развивались обе ветви катализа параллельно, движимые одними и теми же силами.
Что имеется в виду? – Те экономические и социальные причины, которые вызывали появление определенной группы каталитических процессов вне зависимости от того, являются ли они гомогенными или гетерогенными.
Для пояснения - истор. Параллель с великими географическими открытиями.
В катализе можно наблюдать определенные аналогии.
Так, 18-19 века в целом характеризуются бурным развитием промышленности, однако химическая промышленность только начинает зарождаться, востребованы в больших количествах в первую очередь минеральные кислоты , поэтому появляются каталитические процессы получения H2SO4 (и гомогенные, и гетерогенные), затем – процессы получения HNO3.
Конец 19 – начало 20 веков – развитие сельского хозяйства – необходимость в минеральных удобрениях –
Чилийская селитра – далеко и дорого – и как бы в ответ –
Появления габеровского процесса синтеза аммиака и процесса окисления аммиака, т.е. синтеза азотной кислоты.
В это время в промышленности в качестве источника сырья доминирует уголь, который является:
А) источником ацетилена – и появляются процессы каталитических превращений ацетилена;
Б) уголь служит источником синтез-газа (смеси СО+Н2), и появляются процессы на основе этих газов.
30-40-е годы 20-го века:
Германия, активно развивающая военную промышленность и нуждающаяся в горючем для военной техники, отрезана от источников углеводородного сырья. Такая ситуация провоцирует бурный всплеск работ по получению синтетического горючего из альтернативных источников сырья –
- это работы Фишера-Тропша по созданию гетерогенных каталитических процессов
- работы Реппе по созданию гомогенных каталитических процессов.
И, наконец, 50-60-е годы прошлого века – начало бурного развития нефтехимии и химии полимеров –
Циглер и Натта создают катализаторы полимеризации олефинов, появляются процессы получения кислоролсодержащих продуктов при окислении олефинов и т.д.
Фактически можно сказать, что развитие каталитической химии в течение длинного исторического периода определялось вполне определенными экономическими и социальными причинами.
Теории катализа
Самые ранние попытки понять механизмы ускорения реакций катализаторами относятся к началу 19-го века (одновременны со становлением химической науки). С самого начала было установлено, что для катализа необходим непосредственный контакт между реагентами и веществом-катализатором (отсюда термин – контактное действие). При контакте происходит изменение свойств реагентов – их активация. По сути дела все теории пытались объяснить, каким образом катализатор изменяет свойства реагентов, и какие свойства катализатора для этого существенны.
Еще в первой половине 19-го века была обнаружена связь между адсорбцией и гетерогенным катализом. Авторы первых адсорбционных теорий катализа - А. Белани (1824) и М. Фарадей (1833). Чуть позже И. Берцелиус писал, что катализ – это превращение веществ под действием специальной «каталитической силы», присущей катализаторам. Биолог Луи Пастер писал, что «брожение следует отличать от обычных каталитических реакций в мертвой природе, т.к. оно связано с жизнедеятельностью микроорганизмов». Исследования, выполненные позднее, позволили показать, что ферменты, извлеченные из клетки, сохраняют свои каталитические свойства и, следовательно, брожение тоже является примером каталитического процесса.
В середине 19-го века была четко сформулирована теория нестойких промежуточных соединений (Л. Плэйфейр, 1848 г.), развитая и дополненная в конце 19-го начале 20-го века П. Сабатье и др. в виде химической теории катализа. Важную роль в понимании механизма катализа сыграло развитие кинетических методов. В 70-х годах 19-го века появилась серия работ К. Гульдберга, П. Вааге, Л. Вильгельми, Я. Вант-Гоффа (1862-1870), связанных с открытием и применением закона действия масс для изучения кинетики химических, в том числе и каталитических, реакций. После этого стало ясно, что зависимость скорости реакции от концентрации катализатора выглядит часто так же, как зависимости скорости от концентраций реагентов, т.е. катализатор не отличается существенно от других участников реакции. Так обстояло дело в гомогенном катализе. В гетерогенном катализе «неизменность» катализатора в ходе процесса и отсутствие методов фиксации промежуточных соединений стимулировали развитие «нехимических» теорий катализа.
Катализом занимались почти все выдающиеся физики и химики 19-го века. Кроме уже названных, хотелось бы отметить отечественных ученых - Д.П. Коновалова, Н.Д. Зелинского, Д.И. Менделеева.
В начале 30-х 20-го века появилась «Мультиплетная теория» Баландина, в которой наиболее четко формулированы основы гетерогенного катализа, и в первую очередь – геометрические требования к гетерогенному катализатору. В основе этих теорий лежит представление о структуре активного центра на поверхности катализатора. Существование активных центров на поверхности гетерогенных катализаторов было показано в работах Тейлора (1926).
Что имеется в виду?
Геометрические теории придают значение соответствию между геометрической конфигурацией активных атомов на поверхности катализатора (мультиплет) и структурой той части реагирующей молекулы, которая при адсорбции взаимодействует с катализатором. Эту часть молекулы называют индексной группой. Например, для реакций гидрирования алкенов оптимальным оказалось расстояние 2,4-2,8 Ǻ между атомами металла. В этот диапазон попадают параметры кристаллической решетки практически всех металлов 8-й группы Периодической системы. И эти металлы действительно являются лучшими катализаторами гидрирования. Изучение кинетики реакций, протекающих на различных кристаллических гранях металлов, показывает, что скорость действительно зависит от геометрического расположения атомов. Введение дефектов холодной прокаткой металлических пластинок, дроблением или радиоактивной бомбардировкой может менять скорость реакции (особенно при низких температурах, когда дефекты сохраняются). Геометрический подход позволил установить важный факт: селективность в случае конкурирующих реакций может существенно меняться в зависимости от числа и расположения центров. Это привело к развитию представлений об «ансамблях», или специфических группировках атомов на поверхности катализатора, и о структурной чувствительности реакций, скорости которых зависят от размера частиц катализатора (гидрирование – структурно нечувствительная реакция, а гидрогенолиз – структурно чувствительная реакция), возможности образования сплавов и др. факторов.
Полезность такого подхода ограничена в связи с тем, что изменить геометрическое расположение атомов на поверхности катализатора, не меняя при этом какие-либо другие его свойства, очень трудно. И, очевидно, что такое сложное явление как катализ нельзя сводить только к геометрии активных центров. Тем не менее, на сегодня эта теория имеет не только историческое значение и в связи с развитием химии кластеров некоторые положения этой теории получили несколько неожиданное освещение.
(Дать основные определения кластеров, примеры кластеров металлов VIII группы, где действительно часто реалтзуется расстояние 2.4-2.8 А – длина связи металл-металл в металлоостове (например, карбонилфосфиновые кластеры палладия – активные катализаторы гидрирования) показать сходство между ансамблем металлов на поверхности гетерогенного катализатора и металлоостовом кластеров)
Вывод: такие кластерные соединения могут служить моделями активных “ансамблей” на поверхности гетерогенного катализатора
В 50-е годы на волне успехов физики появилась Электронная теория катализа (Ю. Волькенштейн и др.). Согласно этой теории адсорбция и каталитические свойства определяются в первую очередь макроэлектронными свойствами вещества и, в частности, полупроводниковых оксидов. По Волькенштейну, скорость реакции регулируется всей массой имеющихся в катализаторе нелокализованных носителей заряда – электронов или дырок. Влияя добавками (этот процесс называется допирование) на указанные свойства (потенциал ионизации, работа выхода электрона и т.д.) можно изменять каталитические свойства оксидных катализаторов полупроводникового типа. Эта теория имела очень ограниченное применение и в настоящее время практически не используется.
Доминирующим в настоящее время является химический подход (см. выше), дополненный представлениями о важности коллективных факторов (см. ниже) и влиянии носителя в гетерогенном катализе.