Доклад: Значимость острого воспаления в инициировании атеросклероза у детей
Министерство Здравоохранения Республики Узбекистан
Ташкентский Педиатрический Медицинский Институт
Кафедра биологической химии
ДОКЛАД
«Значимость острого воспаления в инициировании атеросклероза у детей»
Сидикова Н.Т., 218 Б группа, педиатрический факультет
Научный руководитель: к. м. н. доц. Хайбуллина З. Р.
Ташкент – 2009г.
План
1. Характеристика ЛПНП.
2. Воспаления – существенная роль в развитии атеросклероза.
3. Белки Острой Фазы способны модифицировать ЛПНП.
4. Действие вирусов, токсинов.
Вывод.
Взаимоотношение воспаления и атеросклероза является темой научной дискуссии более 150 лет. В.Н.Титов (2000) выдвигает гипотезу о ключевой роли в патогенезе атеросклероза дефицита в клетках полиненасыщенных жирных кислот. По мнению Титова, атеросклероз – специфический синдром воспаления, пусковым моментом патогенеза которого является блокада рецепторного поглощения клетками ЛПНП и внутриклеточный дефицит эссенциальных полиненасыщенных жирных кислот (поли ЖК).
Биохимическую основу патогенеза атеросклероза составляет блокада Апо-В100 рецепторного эндоцитоза липопротеинов низкой плотности (ЛПНП). У человека ЛПНП являются основной транспортной формой эссенциальных поли-ЖК в форме эфиров холестерина ( поли – ЭХ), точнее, этерифицированных холестеринов (Хс) эссенциальных поли-ЖК. ЛНП апоВ-рецептор
Различие физико-химических свойств и функциональных особенностей ЛПНП является основой разделения ЛПНП в норме на два субкласса:
ЛПНП-1-большие по размерам, малой плотности липидтранспортные молекулы с еще высоким содержанием ТГ, наличием н-ЭХ, отсутствием поли-ЭХ и пока неактивным положением апоВ-100-лиганда; с ЛПНП-1 ассоциирован апоС-III.
ЛПНП-2 – средние по размерам, более плотные ЛП, с более низким, чем в ЛПНП-1, содержанием ТГ и существенно более высоким уровнем поли-ЭХ, оптимальной конформацией апоВ-100 и активным положением лиганда, который взаимодействует с апоВ-100-рецепторами.
ЛПНП могут быть блокированы белками острой фазы воспаления – амилоидом А, С – реактивным белком, α 1 – ингибитором протеиназ, гаптоглобином, фиброногеном, апо(А). В острой фазе воспаления именно сывороточный амилоид А снижает переход поли ЭХ из ЛПВП в ЛППП и ЛПНП. Кроме того, три из перечисленных белка острой фазы воспаления – СРБ, сывороточный амилоид А (САА) и апо(А) обладают высокой тропностью к липидам и циркулируют в крови в ассоции с разными классами ЛП. Белки острой фазы воспаления СРБ, САА апо(а) при атеросклерозе блокируют физиологические лиганды в ЛПОНП и ЛПНП, становятся их патофизиологическими лигандами и переадресовывают поток насыщенных и поли – жк от высоко дифференцированных клеток, для которых они предназначаются, к клеткам в очаге воспаления. Результатом является дефицит поли – жк в высоко дифференцированных клетках.

1 2 3
1-ЛПНП транспортная форма ЭХ поли-ЖК 2- БОФ- блокатор апоВ-100 рецепторного комплекса 3- клетка,которой эссенциальные поли-ЖК жизненно необходимы.
В условиях блокады апо-В -100 эндоцитоза ЛПНП, циркулурищие в крови, становятся антигенами. Длительная циркуляция ЛПНП и ЛППП, как всех иных белковых макромолекул ( креатинкиназа ЛДГ,АЛТ, АСТ ) вызывает активацию нейтрофилов, которые денатурируют ( модифицируют) макромолекулы ЛПНП и ЛППП путем прикрепления к апо-В 100 сиаловых кислот, усиления перекисного окисления апоВ-100 и поли жк в сложных липидах. В результате денатурации в ЛП формирируются патологические эпитопы, которые позволяют другим иммунокомпетентным клеткам распознавать “ чужеродные. ” макромолекулы ЛПНП и ЛППП и удалять их из кровотока.
Клетки РЭС, моноциты и макрофаги интимы сосудов удаляют ЛПНП и ЛППП из крови путем неспецифического фагоцитоза. На клеточной мембране макрофагов модифицированные ЛПНП и ЛППП взаимодействуют со скважендер - рецепторами ( рецепторами - мусорщиками). При этом атеросклеротическое поражение сосуда формируют макрофаги. Чем выше иммунный статус организма, тем активнее макрофаги поглощают денатурированные ЛП, накапливают их в стенке сосудов, тем активнее развивается атеросклероз.
Моноциты захватывают денатурированные ЛП, проникают в стенку сосуда и дают начало липидным пятнам, а позже - атеросклеротическим бляшкам.
Поглощение макрофагами ЛПНП, ЛППП и формирование пенистых клеток происходит там, где локализованы клетки рыхлой соединительной ткани. Внутренняя пограничная пластина интимы является преградой для диффузии ЛП и миграции макрофагальных клеток. В силу этого формирование пенистых клеток и последующие деструктивные изменения захватывают медию и более глубокие слои стенки сосуда.
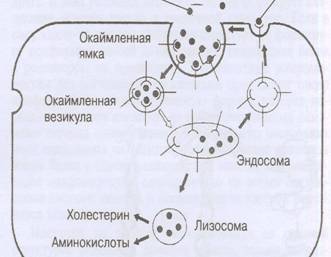
Фагоцитированные макрофагами модифицированные ЛПНП и ЛППП воспринимаются ими как чужые молекулы белка и попадают в лизосомы. В лизосомах кислые протеазы осуществляют протеолиз апо- В 100, но гидролизовать поли -ЭХ на свободную ЖК и свободный ХС ни моноциты ни макрофаги не могут.
Этерифицированные ХС поли - ЖК накапливаются в лизосомах в форме кристаллов поли - ЭХ и превращаются во внутриклеточные инородные тела, которые вначале провоцируют гипертрофию лизосом, а позже кристаллы поли -ЭХ занимают весь цитозоль, формируя пенистые клетки (лаброциты). Далее кристаллы поли- ЭХ механически разрушают лизосомы, вызвая аутолиз и гибель пенистых клеток по типу некроза.
Деструкция клеточных мембран и выход содержимого клеток в межклеточное пространство вызывают реактивные изменения окружающих клеток, гиперпродукцию ими межклеточного матрикса ( коллагена и эластина ) и запускают неспецифичный синдром воспаления. Ситуация усугубляется тем, что в условиях внутриклеточного дефицита поли –ЖК фибробласты и гладкомышечные клетки синтезируют W-9-лейкотриены, которые активируют синдром воспаления и синтез белковых молекул, активирующих миграцию нейтрофилов. При этом вокруг каждой некротизированной клетки формируется очаг активированного асептического воспаления.
Лизис прилежащих клеток, синтез ими интерлейкинов приводят к усилению синтеза гепатоцитами позитивных белков острой фазы : С- реактивного белка, фибриногена, протромбина, апо(а).
С позиций физиологии фагоцитоз клетками денатурированных ЛП и элиминация их из кровотока считается нормальным процессом компенсации. Однако невозможность для макрофагов гидролизовать поли- ЭХ приводит к тому, что процесс компенсации заканчиваются гибелью клеток.
Также, многочисленные микробные токсины способны ингибировать активность ферментов транспорта в крови ЖК. Ингибирование гидролиза Тг в ЛПОНП изменяет поглощение клетками ЛПОНП и ЖК, а ингибирование липолиза в ЛППП и ЛПНП-а нарушает поглощение клетками ЛПНП-б и эссенциальных поли-ЖК. Кроме того, липополисахариды микроорганизмов ассоциируются в крови с ЛПНП и нарушая конформацию апоВ-100, блокируют рецепторное поглощение клетками поли-ЖК. Гепатотоксичное действие микроорганизмов способно ингибировать в гепатоцитах синтез ЛХАТ и блокировать транспорт в клетки поли-ЖК на этапе формирования поли-ЭХ. Действие микробных цитотоксинов способно ингибировать способность гепатоцитами апоА-1 и формирование тех ЛПВП, которые осуществляют отток от клеток свободного Хс. Кроме того, наличие у больного иного хронического заболевания может способствовать развитию атеросклероза, особенно когда заболевание поражает рыхлую соединительную ткань (ревматизм и системная красная волчанка), тем более когда патологический процесс локализован в стенке артерий (болезнь Такаясу).
Внедрение в клетки вирусов существенно действует на процессы клеточной адаптации, изменяет функцию генов, которые кодируют синтез ферментов метаболизма липидов в клетке. Это в большей степени относится к вирусу герпеса и цитомегаловирусам, которые находят в стенке сосуда, особенно в очагах атеросклеротического поражения. В острой фазе восплаения цитомегаловирусы ингибируют активность нейтральной гидролазы моноеновых ЭХ и инициируют накопление их в клетках.
Вирусы способны блокировать в клетках синтез нейтральной гидролазы моно-ЭХ, вызывая накопление в микросомах холестерололеата с формированием пенистых клеток, липидных пятен и полос. Ингибирование гидролиза моно-ЭХ приводит к накоплению их в цитозоле ретикулоэндотелиальных клеток. Возможно, поэтому вирусная инфекция является одним из факторов, который провоцирует формирование рестенозов в коронарных артериях после баллонной дилатации. Аденовирусы индицируют синтез гепатоцитами БОФ, в частности САА, с последующим нарушением как рецепторного поглощения клетками ЛПНП, так и функции апоА-1 ЛПВП. В условиях высокожировой диеты аденовирусы индуцируют гены воспаления и активируют формирование липидной инфильтрации стенки сосудов. При этом формирование липидных пятен и полос из моноеновых эфиров Хс является типичным проявлением острой фазы воспаления.
Вывод
Инфекционные агенты запускают синдром воспаления, БОФ блокируют рецепторный эндоцитоз ЛПНП, что способствует дефициту в клетках ПНЖК, избытку в крови ЛПНП, что трансформирует синдром воспаления в синдром атеросклероза.
БОФ – СРБ, САА, апо(а) – при атеросклерозе как синдроме воспаления блокируют физиологические лиганды в ЛПОНП и ЛПНП, становятся их патофизиологичными лигандами и переадрисуют поток насыщенных и эссенциальных поли-ЖК от высокодифференцированных клеток к клеткам в очаге воспаления.