Курсовая работа: Количественный анализ силибина в экстрактах, полученных с использованием субкритической воды
МИНИСТЕРСТВО ОБРАЗОВАНИЯ И НАУКИ РФ
ГОСУДАРСТВЕННОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ПРОФЕССИОНАЛЬНОГО ОБРАЗОВАНИЯ
«САМАРСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ»
Химический факультет
Кафедра физической химии и хроматографии
Специализация
«химия окружающей среды, химическая экспертиза, экологическая безопасность»
Курсовая работа
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ СИЛИБИНА В ЭКСТРАКТАХ, ПОЛУЧЕННЫХ С ИСПОЛЬЗОВАНИЕМ СУБКРИТИЧЕСКОЙ ВОДЫ
Выполнила студентка
Мелёшкина Екатерина Геннадьевна
Научные руководители
д.т.н., доцент Платонов И.А.
м.н.с. Никитченко Н.В.
Самара 2010
Содержание
Введение
1. Обзор литературы
1.1 Сбор, сушка лекарственных растений и сохранение полученного из них сырья
1.2 Приготовление лекарственных форм из растительного сырья
1.3 Методы анализа лекарственных средств
1.4 Основные направления поиска и создания лекарственных веществ
2. Экспериментальная часть
2.1 Реагенты и оборудование
2.2 Условия выполнения измерений
2.3 Условия безопасного проведения работ
2.4 Методика эксперимента и схема описания установки для экстракции водой в субкритическом состоянии
2.4.1 Подготовка лекарственного растительного материала для экстракции
2.4.2 Методика проведения экстракции субкритической водой
2.5 Подготовка хроматографа к работе и проведение хроматографического анализа
2.6 Проведение градуировки
2.7 Оценка погрешностей измерения определяемых величин
3. Обсуждение результатов
Заключение
Список используемых источников
Введение
Хорошее здоровье – основа долгой, счастливой и полноценной жизни. Чтобы поддерживать здоровье, необходимы знания о свойствах применяемых лекарств, лекарственных растений, а также биологически активных соединениях, входящих в их состав.
Среди лекарственных растений для производства ценных гепатопротекторных препаратов таких, как Силимар, Карсил, Легалон, Силибор и другие широко применяют плоды расторопши пятнистой [Silybum marianum (L.) Gaertn., сем. Asteraceae - Астровых]. Уникальность данных препаратов заключается в том, что их гепатозащитные свойства обусловлены новой группой биологически активных соединений (БАС) флаволигнанами, в частности, силибином [1]. Для производства биологически активных добавок (БАД) и лекарственных препаратов используют зрелые плоды растения, из которых получают экстракты и концентрированные вытяжки флавоноидных фракций растения.
При этом важно подчеркнуть, что гепатопротекторы на основе плодов расторопши пятнистой используются не только для лечения патологии печени, но и в качестве вспомогательной терапии многих инфекционных поражений (хронический бронхит, хламидиоз, токсоплазмоз и др.), а также для профилактики различных заболеваний, возникших в результате воздействия на организм неблагоприятных факторов окружающей среды.
Однако отсутствие на сегодняшний день достаточного ассортимента отечественных аналогов и дороговизна импортных препаратов не позволяют удовлетворять острую потребность населения в данной группе лекарственных средств. Огромные возможности для создания современных гепатопротекторов на основе флаволигнанов расторопши пятнистой имеются в Самарской области, где находится мощная сырьевая база растения.
Для извлечения флаволигнанов из плодов расторопши пятнистой, широко используют органические растворители, большинство из которых токсичны и огнеопасны. Процесс удаления органических растворителей из конечных продуктов существенно влияет на качественный и количественный состав получаемых экстрактов.
Таким образом, разработка отечественных, эффективных, безопасных, доступных новых экстракционных технологий по извлечению биологически активных соединений из сырья лекарственных растений, является актуальной задачей.
Одним из перспективных направлений развития данного метода является экстракция субкритической водой.
В связи с этим, целью данной работы являлось получение «водного» экстракта субкритической водой при температуре 100°С, упаривание его до сухого остатка и оценка растворимости полученного «сухого» экстракта в водной и спиртовой средах.
1. Обзор литературы
1.1 Сбор, сушка лекарственных растений и сохранение полученного из
них сырья
Растительное сырье и лекарства можно получать как из культивируемых, так и из дикорастущих растений. В настоящее время во многих странах значительную часть растительных видов лекарственного сырья получают из культивируемых растений. В Болгарии, за исключением эфирно-масличных культур, главным источником получения сырьевых материалов для фармацевтической промышленности и лекарственных растений являются природные ресурсы этих растений [1]. Однако естественные месторождения их непрерывно уменьшаются или исчерпываются, ввиду чего необходимо направить усилия на получение сырья из культивируемых растений. Известно, что внедренные в культуру растения обладают рядом преимуществ – большим урожаем, более высоким содержанием биологически активных веществ, механизированной уборкой и др. Для получения лекарственного сырья собирают или надземные части всего растения или отдельные его органы. С момента отрывания различных частей растения начинают происходить существенные биохимические изменения. При правильном направлении этих изменений можно получить необходимый состав собранного сырья. В некоторых случаях изменения наступают под воздействием ферментов, особенно во время сушки. Для лекарственного состава в них имеет значение также и время года при проведении сбора и условия сушки [2].
Сбор проводится в светлое и солнечное время и в подходящее время года, вручную или механизированно. При сборе вручную травы, листья и цветки помещают в приспособленные для этого сосуды (корзины, мешки, ящики и др.), следя за тем, чтобы их не сминались и не сдавливались, после чего быстро переносят их в место для сушки. Если транспортировка подготовленного сырья затруднена, то собранные части растений расстилают в закрытых помещениях тонким слоем на полки, пол и др. Там растительный материал может оставаться не дольше 10-12 часов.
Время года, в которое проводится сбор, имеет особое значение. В растении постоянно происходят биохимические процессы и сбор необходимо проводить именно в такой момент, когда в нем сформирован состав с наиболее благоприятным фармакологическим эффектом. Чтобы достичь этой цели, необходимо соблюдать некоторые правила. Так, например, надземные органы (цветки, листья, вся надземная часть) собирают в период цветения растения, а подземные органы (корни, корневища и клубни) - весной, когда вегетация еще не началась или же осенью, когда уже подходит к концу. Перечисленные правила сбора лекарственного сырья, в большинстве случаев, однако, не учитывают особенности отдельных растений и условий, от которых зависит увеличение или уменьшение биологически активных веществ в период одной вегетации. Поэтому, чтобы определить, когда растение находится в «фармакологической зрелости», то есть, когда в них содержится наибольшее количество лекарственных веществ, необходимо проследить в количественном отношении за накоплением веществ во время вегетации. Если лекарственное вещество относится к категории резервных, какими являются, например, слизистые вещества и вообще углеводы, то сырье - подземный орган, естественно, следует собирать осенью, так как в этот конечный период вегетации подземные органы наиболее богаты резервными веществами. Однако, если активный компонент принадлежит к группе вторично образованных веществ, например, алкалоидов, это правило не надо соблюдать [2]. Например, корни красавки в этот период также наиболее богаты крахмалом, но алкалоиды ее содержатся в большем количестве в корнях, собранных до наступления осени (в сравнении с содержанием их в осенних корнях). Этот пример, как и многие другие, показывает, что подходящий для сбора лекарственных растений сезон следует определять с учетом фармакологической зрелости растения. Также имеет значение и в какую часть суток будет собрано лекарственное растение. Известно, например, что биологическая активность растений, содержащих сердечные гликозиды, снижается ночью вследствие распада гликозидов и снова повышается днем при начале ассимиляции, то есть биосинтеза гликозидов. Он достигает максимума во второй половине дня и это время наиболее подходящее для сбора таких растений.
Собранные растения или их органы сразу же после сбора подвергают обработке с целью консервировать, то есть привести их в состояние, во время которого при хранении до промышленной переработки или использования в аптеке в их составе не наступит изменения [3].
Некоторые виды сырья не подвергаются консервированию, так как в них содержатся компоненты, разлагающиеся в ходе этого процесса. Такое сырье перерабатывается в фармацевтические препараты еще в свежем виде.
Некоторое время после сбора растительный орган продолжает жить, хотя и в полностью измененных условиях обмена веществ. Глубокие изменения в тканях начинаются, когда вследствие потери влаги наступило такое состояние увядания, при котором клетки тканей постепенно погибают, то есть перестают быть регулярно функционирующими системами в метаболизме. Содержащиеся в них энзимы, которые уже не принимают участия в биохимических процессах живой ткани, спонтанно катализируют распад лабильных веществ, содержащихся в клетке. Отсюда видно, что изменения, наступающие при превращении свежей части растения в лекарственное сырье, играют огромную роль [4].
Вообще очень трудно или почти невозможно сохранить химический состав и биологическое действие свежих частей растений полностью неизмененными в сырье. Главным образом нужно стремиться при сушке и консервировании не допустить чувствительной потери активных компонентов и сохранить их лечебное действие.
После очистки сырье подвергают высушиванию. Правильное высушивание следует проводить с учетом химизма активных компонентов в сырье. Не допускается высушивание слежавшегося и подверженного ферментации материала. Сушить растительное лекарственное сырье надо быстро при установленной для данного вида и состава сырья температуре. Как показали эксперименты, наиболее подходящей для высушивания сырья является температура около 50°С. При такой температуре действие энзимов ослабевает или полностью прекращается. В некоторых случаях рекомендуют в начале сушку проводить при более высокой температуре, а затем при температуре – около 50°С.
Быстрое высушивание проводят в специальных сушильных камерах, элеваторных сушильнях, вакуумных сушильнях и др., которые оборудованы приспособлениями для регуляции температуры. Особенно быстро необходимо высушивать сочные плоды, содержащие витамины. При этом температуру можно повысить до 70–90°С, благодаря чему значительная часть витаминов сохраняется. Также быстро необходимо сушить и сырье, содержащее сердечные гликозиды, и содержащие алкалоиды сырье. При температуре 50°С и хорошей вентиляции воздуха осуществляется высушивание сырья без изменения его лечебного состава [2, 4].
Медленное высушивание осуществляют на открытом воздухе и в приспособленных для этой цели помещениях. При таком способе высушивания материала хорошие результаты наблюдаются в районах с сухим и теплым климатом. Сырье, которое подвергается сушке, расстилают тонким слоем на деревянных рамках с сетчатым дном, чем обеспечивается более хорошее проветривание при расположении рамок одну над другой во время сушки; сушить следует до тех пор, пока растительные части не станут ломкими и утратят свою эластичность.
Введен метод высушивания лекарственного сырья при инфракрасном свете. При этом инфракрасные лучи проникают внутрь растительного материала, в результате чего процесс высушивания происходит очень быстро. Однако этот метод трудно использовать при больших количествах сырья.
Лекарственное сырье можно сушить и путем лиофилизации. Такой метод применяют при высушивании растительного сырья, активно действующие компоненты которого особенно легко расщепляются. Лиофилизацию проводят при низкой температуре (около 20°С). Содержание влаги в высушенном материале только 2–4,5%. Установлено, что при таком методе высушивания лекарственного сырья, содержащего тропановые алкалоиды, налицо более высокий процент, алкалоидов, чем в сырье, высушенном при 50°С [5].
В процессе сушки осуществляется консервирование сырья, однако не гарантирована его полная стабилизация, вследствие чего некоторые авторы считают, что галеновые препараты (тинктуры, экстракты и др.) – более совершенные формы, в которых лечебный фактор находится в более стойком виде. Теперь применяют и высокоочищенные препараты, которые более стабильны, чем галеновые. В последнее время в Болгарии внедрены в практику и сухие (распыленные) водные экстракты из сырья – дисперги.
После сушки сырье подвергают новой очистке, сортировке, окончательному высушиванию, соответственно измельчению и упаковке. Цель последней очистки – удалить из сырья случайно попавшие в него посторонние части растений или же части, которые во время высушивания утратили свою естественную окраску. Сырье окончательно высушивают, чтобы содержание влажности в нем удовлетворяло требуемому по нормам фармакопеи или стандартам. Нельзя допускать пересушивания, так как такое сырье легко ломается и превращается в порошок (в особенности, листья, цветки) при упаковке и транспорте.
Сортировку проводят согласно указаниям стандартов для различных качеств одного и того же вида сырья (I, II и III) и в зависимости от предназначения – аптечное или промышленное сырье [6].
Физиологически активные вещества растительного происхождения, которые используют непосредственно или в форме трав для лечебных целей, относятся преимущественно к следующим основным химическим группам соединений, как:
1. Алкалоиды.
2. Терпеноиды – монотерпены, сесквитерпены, дитерпены, тритерпены, стеролы, каротеноиды.
3. Производные фенола – фенолы, фенольные кислоты, дубильные вещества, флавоноиды, кумарины, антрахиноны,
4. Углеводы – моносахариды, олигосахариды, полисахариды, сахарные спирты.
5. Глицериды – растительные масла, витамин F, эссенциальные жирные кислоты.
Наряду с представителями, которые недвусмысленно принадлежат к одной из указанных выше химических групп, известно и много растительных веществ, имеющих признаки, характерные для нескольких химических групп; в таком случае одного из них выбирают как основного [1].
1.2 Приготовление лекарственных форм из растительного сырья
Основным процессом, применяемым при приготовлении лекарственных форм из растительного сырья, является экстрагирование. Практически каждый технологический режим, связанный с их производством и, независимо от условий, при которых он протекает, включает экстракцию активно действующих веществ из соответствующего растительного сырья.
Ввиду этого в последние годы теория и практика этого процесса особенно интенсивно разрабатываются в химико-фармацевтической промышленности с учетом некоторых специфических особенностей. Последние связаны прежде всего с предварительными технологическими операциями (сушка, измельчение и др.) и со стабильностью лекарственных веществ, которые в известном смысле, осложняют нормальную регуляцию и оптимизацию этих процессов. В определенной степени предварительная обработка изменяет некоторые свойства растительного сырья, включительно и его химический состав, ввиду создания условий для процессов гидролиза и ферментации, которые чаще всего приводят к уменьшению первоначального биологического эффекта (деструктивные и другие изменения в лекарственном веществе) [4, 7].
Несмотря на это, такая форма обработки растительного сырья безусловно более рациональная, так как благодаря качественно новому их состоянию ускоряются процессы резорбции, исключается возможность лишней нагрузки организма ненужными веществами (растительными клетками) и создаются более хорошие условия для стабилизации и стандартизации вытяжки [1, 4, 8].
Применяемые в современной практике методы экстракции лекарственных веществ из растительного сырья разделяют на две принципиально различные группы
1. Методы экстракции, проводимые при обыкновенной температуре.
2. Методы экстракции, проводимые при повышенной температуре.
По одному из указанных методов получают основные группы галеновых препаратов – экстракты, спиртовые настои, высокоочищенные экстрагированные фитопрепараты (неогаленовые), полифракционные экстракты, водяные вытяжки (инфузы и декокты) и др. Эти методы основаны на некоторых закономерностях, связанных с массообменными процессами, свойствами растительных тканей, физико-химическими свойствами растворителя и веществ, подлежащих экстрагированию.
Под «массообменным процессом» необходимо в самом общем смысле понимать перенос веществ путем диффузии в направлении достижения равновесия в системе (выравнивание концентраций). В частности, при получении производных при экстракции, существенное значение имеет массообмен в системах твердое (сырье) – жидкое (растворитель), жидкое – жидкое (при очистке нативных вытяжек), жидкость – газ (испарение, сушка, конденсация) и др. [8]
Процесс массообмена можно рассматривать в трех аспектах в зависимости от условий его выполнения:
Условия, созданные наличием отдельных фаз и распределением компонентов в них. В сущности, они отражают статические закономерности процесса. В самых общих линиях это распределение и закономерности, которым оно подчиняется, можно выразить, используя распределительный коэффициент между обеими фазами: растворенное вещество в экстрагенте, поглощенном растительным сырьем, и раствор вещества в экстрагенте, обливающем частицы растительного сырья. Следовательно, в данном случае на распределение вещества при равновесном состоянии в основных линиях будут оказывать влияние только обменные соотношения обеих фаз.
Созданные условия для массообмена на граничащей фазовой поверхности, которыми определяются начальная и конечная концентрации [9].
Условия, определяющие скорость процесса, которые выражены коэффициентом массообмена. Знание этих процессов и влияющих на них факторов имеет принципиально важное значение для их оптимизации.
В таком случае основным двигателем является так называемый концентрационный градиент: чем он больше, тем активнее протекают процессы массообмена.
Для приготовления лекарственных форм из растительного сырья применяются следующие способы [10]:
1. Мацерация.
Ее осуществляют в условиях комнатной температуры, предварительно заливая измельченное растительное сырье необходимым количеством экстрактора. Процесс длится различное время (от 15–30 минут до нескольких дней), при этом ежечасно необходимо размешивать систему. Во время мацерации начальная скорость экстракции высокая, затем постепенно уменьшается, пока не наступит равновесие в концентрации растворяющегося вещества внутри в тканях сырья и в экстрагирующем агенте. Выделение раствора из лекарственного сырья (например, путем прессования) не обеспечивает полной экстракции содержащегося в ней компонента. Этот метод простой и не требует специальной аппаратуры.
Скорость процесса можно усилить, проводя дву- или многократную мацерацию, то есть сырье заливают частью растворителя. Вытяжку отделяют путем прессования, а остаток заливают второй частью растворителя. Таким образом обеспечивается двукратно максимальная разница в концентрации активно действующего вещества между сырьем и экстрагирующим фактором. Классическая мацерация осуществляется одним растворителем, чаще всего – смесью этанола и воды. Поэтапная мацерация основывается на смене растворителя, чем облегчается вытяжка различных веществ из сырья в зависимости от их растворимости в воде или этаноле. Сначала сырье заливают всем количеством воды и мацерация длится I–3 дня. Затем добавляют 1/2 этанола и мацерация продолжается до 5 дней, после чего добавляют оставшийся этанол и мацерируют еще 5 дней.
2. Вихревая экстракция
Она введена Melichar (1953) и позднее включена в немецкую фармакопею (DAB–7). Она основана на сокращении времени экстракции путем применения очень интенсивного размешивания. Для этой цели используют миксеры или быстрооборотные мешалки, которые одновременно и измельчают сырье. В результате высокой скорости температура спустя 10 минут повышается, достигая около 40–-45°С. что создает трудности при использовании быстролетучих экстракторов
3. Перколяция (фильтрация через жидкий реагент)
Она позволяет ускорить экстракцию и полное извлечение содержимого сырья. Согласно уравнению Фика. скорость процесса экстракции, то есть количество вещества, которое диффундирует за единицу времени в растворитель, прямопропорционально разницам в концентрации диффундирующего в сырье вещества и растворителем и обратнопропорционально расстоянию между этими двумя фазами. Такого ускорения процесса экстракции можно достичь, создавая условия, чтобы чистый растворитель был в возможно самом близком контакте с каждой частицей сырья и вытеснять находящийся уже там раствор. Это можно осуществить, помещая сырье в колонку, через которую пропускают растворитель.
Перколяция осуществляется в 4 этапа:
Мацерация сырья в целях его набухания.
Заполнение перколятора, таким образом, чтобы получить равномерно расположенные капилляры.
Заливание сырья растворителем с одновременным вытеснением воздуха между частицами сырья и последующая мацерация в течение 24 часа.
Перколяция. Заливание сырья жидкостью происходит после его измельчения и смешивания с растворителем, количество которого составляет 40–50% массы сырья; сосуд закрывают и оставляют неподвижным на 2–3 часа. Для увлажнения используют тот же растворитель, которым проводили экстракцию или другие смеси с добавкой кислоты, глицерина или других, облегчающих экстракцию, веществ. На дно перколятора помещают слой ваты. В крупных перколяторах на дне имеется сито или ситовая прокладка, которую покрывают слоем марли. Сырье раскладывают плотными слоями, слегка надавливая, но так, чтобы между ними не было свободного пространства, а также не допуская и чрезмерного слеживания, так как при дополнительном набухании сырья проникание растворителя будет затруднено. На верхнюю поверхность сырья помешают круг фильтровальной бумаги и ситовую прокладку. Следует заполнять сырьем 3/4 объема перколятора. Растворитель заливают в перколятор при открытом кране в нижней части перколятора. В тот момент, когда появляется первая капля растворителя, кран закрывают и доливают такое количество растворителя, чтобы образовался слой около 2–3 см над сырьем. Перкулятор закрывают и выдерживают 24 часа. По истечении срока мацерации кран открывают и скорость вытекания зависит от количества сырья. На каждый кг при получении тинктуры в отношении 1:5–30 капель/мин (0,5–0,6), 1:10–60 капель/мин (1,0–1,2). Одновременно доливают чистого растворителя. Экстракцию путем перколяции проводят или до сбора определенного количества вытяжки (тинктуры), или до полного истощения растительного сырья.
4. Реперколяция
Это метод получения жидких вытяжек при минимальном расходе растворителя и без концентрации. Принцип этого метода основан на прохождении растворителя, содержащего определенное количество экстрагированного вещества, через свежее сырье. Сырье делят на 3 порции: 50, 30, 20 частей. Первую порцию (50) смачивают растворителем и закладывают в перколятор и спустя 24-х часовую мацерацию собирают 20 частей (первая вытяжка) и 5 порций по 30 частей (вторичная вытяжка). Добавляют также и вытяжку, полученную при прессовании сырья. Вторую порцию сырья (30 частей) смачивают частью из порций при вытяжке и подвергают перколяции, последовательно добавляя порции вторичной вытяжки, полученные из первой порции сырья, Первые 30 частей сливают отдельно как часть готовой вытяжки, а следующие порции собирают в 4 очередных порции по 20 частей. С этими порциями поступают также, собирая последние 50 частей вытяжки.
5. Дигестия
Идет речь об экстракции при температуре 30^40'С, а иногда при 50*С.
6. Приготовление инфуэов и отваров
По этим методам приготовляют так называемые настои и отвары. Это водные экстракты из растительного сырья или водные растворы специально полученных для данной цели экстрактов.
Галеновые препараты, наряду с активно действующими веществами, содержат также и ряд сопутствующих веществ, называемых баластными. Роль последних – весьма противоречива. С технологической точки зрения – они нежелательны, так как затрудняют процесс экстракции, отягощают состав полученных вытяжек, становятся причиной появления опалесценции, осадков, вступают во взаимодействие с некоторыми лекарственными средствами и др. С другой стороны, они в состоянии повлиять на резорбцию и на терапевтический эффект активно действующих веществ вытяжки. В литературе имеются данные о более высоком терапевтическом эффекте тотальных вытяжек из растительного сырья в сравнении с изолированными очищенными, активно действующими веществами.
В процессе экстракции растительное сырье в зависимости от механической структуры абсорбирует и задерживает определенное количество экстрактора [7, 11]. Для регламентации этого количества ГФ X вводит показатель «коэффициент поглощения воды» (количество воды, задерживаемое 1 г сырья). Этот коэффициент используют для определения дополнительного количества воды при приготовлении вытяжек.
Для приготовления настоев и отваров растительное сырье необходимо измельчить: листья, цветки, стебли — до размеров, не больше 5 мм (Fol. Uvae ursi и другие виды сырья с кожистыми листьями до 1 мм); стебли, кору, корни, корневища – размерами, не более 3 мм; плоды и семена — не крупнее 0,5 мм.
То время, когда сырье находится в контакте с экстрагирующим веществом называют контактным временем. Для настоев это время равно 60 мин (15 мин нагревания и 45 мин охлаждения), а для отваров – 40 мин (30 мин нагревания и 10 мин охлаждения).
При приготовлении настоев по рецептам с обозначением «Cito» время нагревания увеличивают на 25 мин и затем искусственно охлаждают. Когда готовят водные экстракты в количестве 1000–3000 г, время нагревания соотв. увеличивают: для настоев до 25 мин, а для отваров – до 40 мин [12].
По истечении контактного времени водные экстракты процеживают через сложенную в несколько слоев марлю, вату или их сочетание, выжимают остаток растительного материала и, если масса не соответствует прописанной в рецепте, доливают водой.
Отвары из Fol. Uvae ursi, Cortex Quercus, Cortex Chinae процеживают сразу же после снятия инфундирки с водяной бани, ввиду того, что активно действующие вещества в тепле обладают более высокой растворимостью. Отвары из Fol. Sennae процеживают сразу же после их полного охлаждения.
При приготовлении настоев и отваров путем разведения экстрактов последние следует брать в количестве, соответствующем указанному в рецепте для данного лекарственного сырья [13].
Независимо от указанных трудностей, связанных с приготовлениям фитопрепаратов, практика показывает, что применение их значительно расширяется и их актуальность в настоящее время особенно велика.
1.3 Методы анализа лекарственных средств
Одна из наиболее важных задач фармацевтической химии – это разработка и совершенствование методов оценки качества лекарственных средств.
Для установления чистоты лекарственных веществ используют различные физические, физико-химические, химические методы анализа или их сочетание. ГФ предлагает следующие методы контроля качества ЛС [14].
Физические и физико-химические методы. К ним относятся: определение температур плавления и затвердевания, а также температурных пределов перегонки; определение плотности, показателей преломления (рефрактометрия), оптического вращения (поляриметрия); спектрофотометрия – ультрафиолетовая, инфракрасная; фотоколориметрия, эмиссионная и атомно-абсорбционная спектрометрия, флуориметрия, спектроскопия ядерного магнитного резонанса, масс-спектрометрия; хроматография – адсорбционная, распределительная, ионообменная, газовая, высокоэффективная жидкостная; электрофорез (фронтальный, зональный, капиллярный); электрометрические методы (потенциометрическое определение рН, потенциометрическое титрование, амперометрическое титрование, вольтамперометрия).
Кроме того, возможно применение методов, альтернативных фармакопейным, которые иногда имеют более совершенные аналитические характеристики (скорость, точность анализа, автоматизация). В некоторых случаях фармацевтическое предприятие приобретает прибор, в основе использования которого лежит метод, еще не включенный в Фармакопею (например, метод рамановской спектроскопии – оптический дихроизм). Иногда целесообразно при определении подлинности или испытании на чистоту заменить хроматографическую методику на спектрофотометрическую. Фармакопейный метод определения примесей тяжелых металлов осаждением их в виде сульфидов или тиоацетамидов обладает рядом недостатков. Для определения примесей тяжелых металлов многие производители внедряют такие физико-химические методы анализа, как атомно-абсорбционная спектрометрия и атомно-эмиссионная спектрометрия с индуктивно связанной плазмой [8, 15].
Важной физической константой, характеризующей подлинность и степень чистоты ЛС, является температура плавления. Чистое вещество имеет четкую температуру плавления, которая изменяется в присутствии примесей. Для лекарственных веществ, содержащих некоторое количество допустимых примесей, ГФ регламентирует интервал температуры плавления в пределах 2 °С. Но в соответствии с законом Рауля (AT = iK3C, где AT – понижение температуры кристаллизации; К3 – криоскопическая постоянная; С – концентрация) при i = 1 (неэлектролит) значение АТ не может быть одинаковым для всех веществ. Это связано не только с содержанием примесей, но и с природой самого ЛВ, т. е. с величиной криоскопической постоянной К3, отражающей молярное понижение температуры плавления ЛВ. Таким образом, при одинаковом AT = = 2 "С для камфоры (К3 = 40) и фенола (К3 = 7,3) массовые доли примесей не равны и составляют соответственно 0,76 и 2,5 %.
Для веществ, которые плавятся с разложением, обычно указывается температура, при которой вещество разлагается и происходит резкое изменение его вида.
Критериями чистоты являются также цвет ЛВ и/или прозрачность жидких лекарственных форм [16].
Определенным критерием чистоты ЛС могут служить такие физические константы, как показатель преломления луча света в растворе испытуемого вещества (рефрактометрия) и удельное вращение, обусловленное способностью ряда веществ или их растворов вращать плоскость поляризации при прохождении через них гаюскополяризованного света (поляриметрия). Методы определения этих констант относятся к оптическим методам анализа и применяются также для установления подлинности и количественного анализа ЛС и их лекарственных форм.
Важным критерием доброкачественности целого ряда ЛС является содержание в них воды. Изменение этого показателя (особенно при хранении) может изменить концентрацию действующего вещества, а, следовательно, и фармакологическую активность и сделать ЛС не пригодным к применению [17].
Химические методы. К ним относятся: качественные реакции на подлинность, растворимость, определение летучих веществ и воды, определение содержания азота в органических соединениях, титриметрические методы (кислотно-основное титрование, титрование в неводных растворителях, комплек-сонометрия), нитритометрия, кислотное число, число омыления, эфирное число, йодное число и др.
Биологические методы. Биологические методы контроля качества ЛС весьма разнообразны. Среди них испытания на токсичность, стерильность, микробиологическую чистоту.
Для проведения физико-химического анализа полупродуктов, субстанций лекарственных средств и готовых лекарственных форм при проверке их качества на соответствие требованиям ФС контрольно-аналитическая лаборатория должна быть оснащена следующим минимальным набором оборудования и приборов:
1. ИК-спектрофотометр (для определения подлинности);
2. спектрофотометр для спектрометрии в видимой и УФ-области (определение подлинности, количественное определение, однородность дозирования, растворимость);
3. оборудование для тонкослойной хроматографии (ТСХ) (определение подлинности, родственных примесей);
4. хроматограф для высокоэффективной жидкостной хроматографии (ВЭЖХ) (определение подлинности, количественное определение, определение родственных примесей, однородности дозирования, растворимости);
5. газожидкостной хроматограф (ГЖХ) (содержание примесей, определение однородности дозирования);
6. поляриметр (определение подлинности, количественное определение);
7. потенциометр (измерение рН, количественное определение);
8. атомно-абсорбционный спектрофотометр (элементный анализ тяжелых металлов и неметаллов);
9. титратор К. Фишера (определение содержания воды);
10. дериватограф (определение потери массы при высушивании).
1.4 Основные направления поиска и создания лекарственных веществ
Создание лекарственного препарата – длительный процесс, включающий несколько основных этапов – от прогнозирования до реализации в аптеке [3, 18].
В создании новых ЛС участвуют представители многих профессий: химики, биологи, фармацевты (провизоры), фармакологи, токсиколога, врачи-клиницисты. Однако совместные усилия специалистов не всегда завершаются успешно. Из мировой фармацевтической практики следует, что из 10 тыс. вновь синтезированных органических соединений только одно может использоваться как лекарственное средство [19].
Основой прогнозирования биологической активности лекарственного вещества является установление связи между фармакологическим действием (биологической активностью) и структурой с учетом физико-химических свойств лекарственного вещества и биологических сред.
лекарственный растение экстракт силибин
2. Экспериментальная часть
Экспериментальная часть работы посвящена получению «водного» экстракта субкритической водой при температуре 100°С, упаривание его до сухого остатка и оценка растворимости полученного «сухого» экстракта в водной и спиртовой средах.
2.1 Реагенты и оборудование
При выполнении измерений использовалось следующие средства измерения, оборудование и реактивы:
· Весы лабораторные общего назначения по ГОСТ 24104 с наибольшим пределом взвешивания 200 г, 2 класса точности
· Набор гирь Г-2-200 - ГОСТ 7328
· Экспериментальная установка для ЭСВ
· Плоды расторопши пятнистой
· Высокоэффективный жидкостный хроматограф «Biotronik» со спектрофотометрическим детектором λ=190-700нм
· Хроматографическая колонка из нержавеющей стали фирмы «Phenomenex», USA, длиной 250 мм, диаметром 4,6 мм с неподвижной фазой С18, зернением 5 мкм
· Кислота соляная - ТУ 6-09-2540-87
· Вода дистиллированная - ГОСТ 6709
· Метанол, х.ч. - ГОСТ 6995
· ГСО силибина
· Ацетонитрил «extra pure» фирмы «MERCK»
· Калий фосфорнокислый однозамещенный, ч.д.а. - ТУ 6-09-5324-87
· Универсальные индикаторные бумаги рН=0-12
· Гелий газообразный - ГОСТ 9093-74
· Мерная колба вместимостью 1000 см3
· Цилиндры мерные 2-100, 2-500 – ГОСТ 1770-74
· Шприцы медицинские 1 мл, 5 мл
· Микропипетки на 200 мкл – ГОСТ 29169
· Микрошприц фирмы «Hamilton» на 100 мкл.
2.2 Условия выполнения измерений
· Температура окружающей среды(20 ± 5)°C;
· Относительная влажность(80 ± 5) %;
· Атмосферное давление(94 – 106) кПа;
· Частота переменного тока(50 ± 1) Гц;
· Напряжение в сети(220 ± 10) В.
2.3 Условия безопасного проведения работ
При выполнении анализов необходимо соблюдать требования техники безопасности при работе с химическими реактивами по ГОСТ 12.4.021, электробезопасность при работе с электроустановками по ГОСТ 12.1.019, организации обучения работающих безопасности труда по ГОСТ 12.0.004. Помещение лаборатории должно соответствовать требованиям пожарной безопасности по ГОСТ 12.1.004 и иметь средства пожаротушения по ГОСТ 12.4.009.
2.4 Методика эксперимента и схема описания установки для
экстракции водой в субкритическом состоянии
С целью постановки эксперимента по получению экстрактов плодов расторопши при температуре 100°С и давлении 0,5 МПа была сконструирована и смонтирована установка для экстракции водой в субкритическом состоянии, схема которой представлена на рисунке 1.
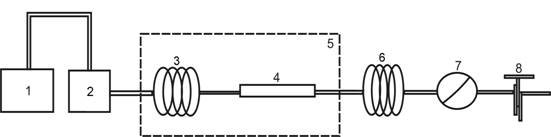
Рис. 1. Схема установки для экстракции субкритической водой:
1 - сосуд с водой; 2 - насос высокого давления; 3 - капилляр предварительного нагрева воды; 4 - экстрактор; 5 - термостат; 6 - охлаждаемый капилляр; 7 - манометр; 8 - регулятор давления
В данной установке дистиллированная дегазированная вода из сосуда 1 подается в насос высокого давления 2, обеспечивающий расход в системе от 0,1 до 10,0 см3/мин при давлении до 40,0 МПа. Из насоса 2 вода поступает в капилляр предварительного нагрева воды 3 и экстрактор 4, помещенные в термостат 5. Экстрактор 4 выполнен в виде колонки из нержавеющей стали длиной 250 мм и внутренним диаметром 10 мм. На выходе из экстрактора 4 установлен капилляр 6, охлаждаемый до Т = 20°С. Для контроля давления в системе установлен образцовый манометр 7 и регулятор давления 8.
2.4.1 Подготовка лекарственного растительного материала для
экстракции
На основании анализа литературных источников было установлено, что максимальное содержание биологически активных соединений находится в плодах расторопши пятнистой, причем более 95% этих соединений содержится в лузге.
Кроме того, выбор лузги семян расторопши пятнистой с зернением 0,1-0,3 мм обусловлен тем, что при этом обеспечивается хорошая проницаемость подвижной фазы в экстракторе, заполненном лузгой.
Для проведения эксперимента по экстракции была использована фракция лузги расторопши пятнистой, приготовленная из одной партии плодов растения, предоставленной ЗАО «Самаралектравы» (с. Антоновка, Самарская область).
Приготовление лузги расторопши пятнистой осуществлялось следующим образом: проводили измельчение плодов, затем отсеивали лузгу от муки семян расторопши пятнистой на почвенных ситах диаметром ячейки 0,1 – 0,5 мм.
2.4.2 Методика проведения экстракции субкритической водой
Заполнение экстракционного сосуда лузгой семян расторопши пятнистой проводили методом сухого заполнения колонны экстрактора, предварительно на одну часть колонны устанавливали фитинги и фритты. На заполнение экстракционного сосуда требовалось в среднем 11,4 г навески лузги. Затем на открытую часть колонны экстрактора также устанавливали фитинги и фритты. Экстрактор устанавливали в термостат, заполняли подвижной фазой и устанавливали требуемые для опыта температуру и давление.
Затем в течение 20-30 мин выдерживали систему в статическом режиме, после чего процесс экстракции проводили динамическим способом в течение 40-60 мин при расходе подвижной фазы 1 мл/мин, отбирая по 5мл в коллектор фракций с последующим их анализом методом обращено-фазовой высокоэффективной жидкостной хроматографией.
2.5 Подготовка хроматографа к работе и проведение
хроматографического анализа
Приготовление подвижной фазы
Для проведения измерений методом ВЭЖХ готовили смесь ацетонитрила с 0,01М раствором фосфатного буфера в объемном соотношении 35:65. Требуемые объемы ацетонитрила и фосфатного буферного раствора отмеряли мерными цилиндрами.
Для приготовления подвижной фазы использовали ацетонитрил «extra pure» фирмы «MERCK».
Приготовление раствора фосфатного буфера.
0,1М раствор фосфатного буфера готовился путем растворения 13,8 г натрия фосфорнокислого (NaH2PO4·H2O) в 900 см3 бидистиллированной воды, раствор тщательно перемешивали и доводили до 1000 см3. Затем 100 см3 полученного 0,1М буферного раствора разбавляли водой до 1000 см3 и добавляли 2М раствор соляной кислоты до рН=3.
Готовую подвижную фазу дегазировали гелием.
Режим работы хроматографа для определения времени удерживания исследуемых компонентов:
· Температура колонки25оС;
· Скорость потока0,6 мл/мин;
· Объем вводимой пробы20 мкл;
· Длина волны 289 нм
· Время анализа 20 мин.
2.6 Проведение градуировки
Качественный анализ компонентного состава экстрактов проводили по временам удерживания БАС относительно времени удерживания стандартного образца сравнения (силибина) с использованием литературных данных [20, 21].
Количественный анализ силибина проводили методом абсолютной градуировки.
Приготовление градуировочных смесей
Построение градуировочных характеристик (зависимость площади хроматографического пика вещества от его концентрации) осуществляли с использованием государственного стандартного образца (ГСО) силибина (рег. №42-0072-01).
Для приготовления градуировочных растворов силибина 0,02 г ГСО силибина растворяли в мерной колбе вместимостью 100 мл в 80 мл 95%-ного этилового спирта при нагревании (Т=70°С), затем содержимое колбы охлаждали и доводили объем раствора до метки. Далее объемно-весовым методом готовили серию градуировочных растворов силибина в этаноле (0,5 – 50 мг/дм3).
Значения концентраций таксифолина, силикристина и силидианина рассчитывали методом внутреннего стандарта по уравнению:
![]() ,(1)
,(1)
где ![]() - площадь
хроматографического пика определяемого компонента;
- площадь
хроматографического пика определяемого компонента; ![]() -
площадь хроматографического пика стандарта (силибина);
-
площадь хроматографического пика стандарта (силибина); ![]() - концентрация стандарта.
- концентрация стандарта.
Коэффициенты чувствительности
для исследуемых компонентов относительно стандартного вещества силибина равны
единице, так как все они имеют одинаковое поглощение при длине волны
спектрофотометрического детектора ![]() = 289 нм
[1].
= 289 нм
[1].
Построение градуировочных зависимостей
По полученным результатам пяти параллельных измерений для каждого градуировочного раствора стандарта (силибина) были построены градуировочные характеристики, описываемые уравнением
![]() ,(2)
,(2)
где ![]() и
и ![]() - входной и выходной
сигналы хроматографа соответственно,
- входной и выходной
сигналы хроматографа соответственно, ![]() ‑концентрация
компонента в градуировочной смеси;
‑концентрация
компонента в градуировочной смеси; ![]() –
площадь хроматографического пика;
–
площадь хроматографического пика; ![]() -
константа корреляционного уравнения, рассчитываемая методом наименьших
квадратов [20]:
-
константа корреляционного уравнения, рассчитываемая методом наименьших
квадратов [20]:
 ,(3)
,(3)
где m=5 –количество градуировочных
растворов; ![]() - среднее арифметическое i-х значений выходного сигнала для
соответствующей концентрации
- среднее арифметическое i-х значений выходного сигнала для
соответствующей концентрации ![]() в
растворе.
в
растворе.
2.7 Оценка погрешностей измерения определяемых величин
По результатам анализа градуировочных растворов силибина проведена оценка правильности и прецизионности измерения [22]. Для этого рассчитывали абсолютный коэффициент чувствительности для расчета концентрации силибина в исследуемых смесях:
![]() ,(4)
,(4)
где ![]() - измеренная концентрация
силибина в i-ом градуировочном растворе,
- измеренная концентрация
силибина в i-ом градуировочном растворе, ![]() - абсолютный коэффициент
чувствительности для силибина или тангенс угла наклона зависимости
- абсолютный коэффициент
чувствительности для силибина или тангенс угла наклона зависимости ![]() .
.
1. Правильность измерения, ![]() (%) в каждой выборке
определяли по уравнению
(%) в каждой выборке
определяли по уравнению
![]() (5)
(5)
где ![]() - среднее значение
измеренной концентрации в i-ом
градуировочном растворе,
- среднее значение
измеренной концентрации в i-ом
градуировочном растворе, ![]() -
истинная концентрация силибина в градуировочном растворе.
-
истинная концентрация силибина в градуировочном растворе.
2. Прецизионность измерения:
- Среднее квадратичное отклонение единичного измерения концентрации в i-ой выборке:
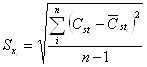 ,(6)
,(6)
где n=5 – число измерений в выборке.
- Граница доверительного интервала измерений для i-го градуировочного раствора:
![]() ,(7)
,(7)
где ![]() - критерий Стьюдента при
доверительной вероятности P=0,95
и числе степеней свободы f=n-1.
- критерий Стьюдента при
доверительной вероятности P=0,95
и числе степеней свободы f=n-1.
- Предел прецизионности ri в условиях повторяемости для двух измерений:
![]() ,(8)
,(8)
где 1,96 – коэффициент критического диапазона при Р=0,95.
- Относительное среднее
квадратическое отклонение (ОСКО) среднего арифметического результата измерения ![]() (%) в выборке:
(%) в выборке:
![]() (9)
(9)
В таблице 1 приведены показатели точности измерения концентрации силибина в градуировочных растворах.
Таблица 1. Показатели точности определения концентрации силибина
|
Истинная концен-трация
|
Измерен-ная концентра-ция
|
Разность двух концентраций в выборке
|
Предел прецизион-ности ri, мг/дм3 |
Граница доверительного интервала
|
Правиль-ность измере-ния
|
ОСКО
|
| 0,50 | 0,45 | 0,09 | 0,10 | 0,05 | 10,00 | 3,70 |
| 1,00 | 0,89 | 0,14 | 0,20 | 0,08 | 11,00 | 3,00 |
| 5,00 | 4,66 | 0,76 | 0,80 | 0,39 | 6,80 | 2,80 |
| 10,00 | 9,48 | 1,77 | 1,90 | 0,89 | 5,20 | 3,20 |
| 50,00 | 45,84 | 6,50 | 7,70 | 3,75 | 8,30 | 2,70 |
Как видно из
данных таблицы 1, разность двух измеренных концентраций силибина (максимальной
и минимальной) для всех градуировочных растворов не превышает предела
прецизионности ri,, а общая погрешность
измерения концентрации силибина с учётом правильности и прецизионности может
быть принята на уровне ![]() относительно
измеряемой концентрации.
относительно
измеряемой концентрации.
3. Обсуждение результатов
Создание и производство лекарственного препарата, как на основе синтезированных форм, так и на основе лекарственных растений – задача сложная. Наряду с технологическими и производственными задачами в первую очередь возникают проблемы, связанные с получением фармацевтических субстанций с особой химической чистотой, качеством и терапевтической эффективностью. В то же время лекарственные формы должны отвечать определенным требованиям, одним из которых является растворимость. Очевидно, что размер частиц определяет размеры поверхности, которые в свою очередь контролируют скорость растворения и действие лекарства. Микро- и наноформы фармпрепаратов обладают уникальными свойствами и преимуществами, открывающими новые перспективные подходы к терапии самых различных заболеваний. Прежде всего, микронизация позволяет существенно повысить скорость растворения гидрофобных фармпрепаратов в водных средах. Также известно, что для достижения терапевтического эффекта малорастворимые в водной среде лекарственные препараты должны применяться в высоких дозах, что обусловливает их нежелательное побочное действие, представляющее серьезную проблему в случае сильнодействующих противоопухолевых, гормональных, противовоспалительных, противогрибковых препаратов и антибиотиков.
В связи с этим в настоящей работе были проведены эксперименты по количественному определению силибина в экстрактах плодов расторопши, полученные субкритической водой при температуре 100°С и давлении 0,5 МПа, а также исследования по эффективности растворения сухого остатка полученного экстракта в водной и спиртовой средах.
Использование субкритической воды для получения экстракта лекарственного растения обусловлен ее эффективностью, экологической безопасностью и экономичностью. Выбор расторопши пятнистой в качестве объекта исследования неслучаен. Не безызвестен тот факт, что основная часть гепатопротекторных препаратов изготовлены из флаволигнанов, которые содержатся в расторопше пятнистой. К ним относятся таксифолин, силикристин, силидианин и силибин [23]. Наибольшую биологическую активность по гепатозащитным свойствам из которых проявляет силибин.
Анализ экстрактов расторопши пятнистой проводили методом высокоэффективной жидкостной хроматографии.
Для оценки растворимости компонента в водной и спиртовой средах и его возврата в исходную субстанцию был проведен ряд стадий эксперимента:
- 5 см3 экстракта расторопши пятнистой, полученного при Т = 100°С и Р = 0,5 МПа, представляющего собой «водный экстракт» упаривали до сухого остатка (было подготовлено два образца сухого остатка);
- полученный «сухой экстракт» растворяли в 5 см3 горячей воды (Т = 100°С и Р = 0,1 МПа) и в 5 см3 метилового спирта, и затем хроматографировали.
На рисунке 2 представлена диаграмма, показывающая концентрацию исследуемого компонента в исходном экстракте, и в экстрактах, полученные путём растворения сухого остатка исходного экстракта в воде и метиловом спирте.
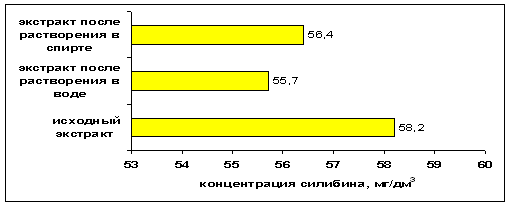
Рис. 2. Диаграмма концентрации силибина (мг/дм3) в экстракте расторопше пятнистой, и в экстрактах, полученных путем растворения сухого остатка экстракта в воде и метиловом спирте
Как видно из рисунка 2 концентрация силибина в экстракте, полученном субкритической водой при температуре 100°С и давлении 0,5 МПа составляет 58,2 мг/дм3. В то же время после получения сухого остатка данного экстракта и растворения его в водной и спиртовой средах, концентрации изучаемого компонента практически сопоставимы. Таким образом, следует судить о том, что переход из «водного экстракта» в «сухой», что является наиболее выгодной формой фармацевтической субстанции, не влияет на изменение качественного и количественного состава экстракта, а, следовательно, и на эффективность лечебного действия получаем лекарственных форм.
Заключение
Проведен анализ литературных источников по способам подготовки лекарственного растительного сырья для производства фармпрепаратов.
Проведен количественный анализ силибина в экстрактах плодов расторопши, полученных субкритической водой при 100°С и давлении 0,5 МПа.
Экспериментально установлено, что концентрации силибина в исходном экстракте, водном и спиртовом растворах (после предварительного высушивания исходного экстракта при Т=40°С и последующего его растворения в них) практически совпадают.
Список используемых источников
1. Акопов И. Заготовка и сушка сырья лекарственных растений // Научно-культурологический журнал. №11. 65. 2001.
2. Государственная фармакопея СССР. 11 изд. Выпуск 2. общие методы анализа. Лекарственное растительное сырьё. Москва: Медицина, 1990. С. 47-148.
3. Козлов А.Т., Артюховский А.К. / Лекарственные растения: Учеб.пособие. – Воронеж : Воронеж. гос. лесотехн. акад. 1999. 175с.
4. Кузнецова М.А. Лекарственные растения и препараты. 1987. Изд. 2.
5. Тюренкова И.Н. Растительные источники витаминов. Волгоград. 1999.
6. Голубев Л.Г., Сажин Б.С., Валашек Е.Р. / Сушка в химико-фармацевтической промышленности/ Медицина, М.: 1978.
7. Новые возможности высокоэффективной жидкостной хроматографии в анализе лекарственных средств // Фарматека. 2002. № 11 (62). С. 71- 74.
8. Гацура В.В. Методы первичного фармакологического исследования биологически активных веществ. М.: Медицина, 2000.
9. Арзамасцев А.П., Печенников В.М., Родионова Г. М. и др. Анализ лекарственных смесей. М.: Спутник, 2000. 275с.
10. Беликов В.Г. Методы анализа. // Фармация. 2000. Т.49. №1. С.23-25.
11. Н.П. Максютина, Ф.Е. Каган, Л.А. Кириченко и др. Методы анализа лекарств. // Киев: Здоровье. 2004. 222с.
12. Краснюк И.Н. Фармацевтическая технология: Технология лекарственных форм. М.: Издательский центр «Академия», 2004.
13. Милованова Л.Н. Разработка из готовления лекарственных форм. Ростов на Дону: Медицина, 2002 - 448 с.
14. Шишков А.Н. Растительные масла и масляные экстракты: технология, стандартизация, свойства. М. Русский врач, 2004.
15. И.И. Перевозченко и др. Лекарственные растения. К.: Урожай, 1991.
16. L. Tripathi, J.N. Tripathi. Role of biotechnology in medicinal plants./ //Tropical Journal of Pharmaceutical Research. 2003. 2. p. 243-253.
17. Birch RG. Plant transformation: Problems and strategies for practical application.// Ann Rev Plant Physiol Plant Mol Biol. 1997. 48. p. 297-326.
18. Stafford A, Morris P, Fowler MW. Plant Cell Biotechnology: A perspective. / Enzyme Microbial Tech. 1986. 8. p. 578-97.
19. Mainchin B., Kettish P., Knapp G. // Fresenius J. Anal. Chem. 2000. 366. P. 26.
20. G. Tittel, H. Wagner. // J. Chromatogr. 1978. - №158. - P. 227-232.
21. Минахметов Р.А., Онучак Л.А., Куркин В.А. // Химия природных соединений.2001. - Т. 37, №4. - С. 318-321.
22. ГОСТ Р ИСО 5725-2002. Точность (правильность и прецизионность) методов и результатов измерений.
23. Залепугин Д.Ю., Тилькунова Н.А., Чернышова И.В. // Журн. сверхкритические флюиды. Теория и практика. 2002. Т.3. №1. С. 5.