Реферат: Библиотека структурных гетероциклических аналогов, содержащих имидный и сульфонильный фрагменты
М. В. Дорогов, Л. А. Савватеева, И. В. Тюнёва
В последние годы проводится все больше исследований, связанных с разработкой методов синтеза органических соединений, обладающих определёнными типами биологической активности и являющихся разнообразными лекарственными препаратами [1,2]. Согласно литературным данным, предпочтение в этих исследованиях отдается гетероциклическим системам, содержащим атомы кислорода, серы, азота и широкое разнообразие функциональных заместителей [3-5]. Одна из причин использования гетероциклических соединений - это широкие возможности их структурной модификации, а, следовательно, получение соединений с новым комплексом биохимических свойств. Одним из вариантов модификации гетероциклических структур является введение различных фрагментов и функциональных групп в качестве заместителей. Поэтому для современной медицинской химии особый интерес представляют комбинаторные библиотеки структурных аналогов с однотипным гетероциклическим скелетом и варьирующимися фрагментами и функциональными группами.
Целью данной работы являлось генерирование библиотеки структурных аналогов гетероциклического типа, содержащих одновременно имидный и сульфонильный фрагменты, идентификация синтезированных соединений и компьютерная оценка их биологической активности с помощью системы PASS [6,7].
Известно, что оба вышеупомянутых структурных фрагмента используются в направленном поиске биологически активных препаратов. Так, в частности, известен имидосодержащий препарат СЕДИЕЛ(r), являющийся эффективным антидепрессантом, а ароматические сульфокислоты считаются перспективными билдинг-блоками для получения различных химиотерапевтических средств [8,9].
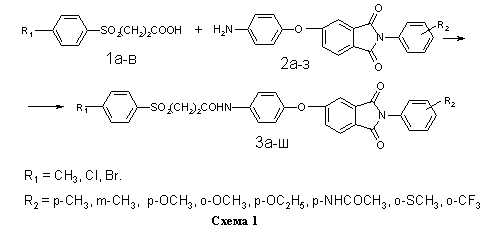
На схеме 1 представлена реакция получения библиотеки структурных аналогов с имидным и сульфонильным фрагментами 3а-ш. В качестве реагентов были использованы ароматические сульфопропионовые кислоты 1а-в и аминофенокси-N-фенилфталимиды 2а-з (таблица).
Соединения 1a-в получали на основе толуола, хлор- и бромбензола по методикам, изложенным в работах [10,11] (схема 2).
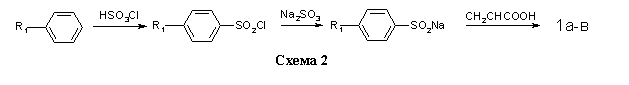
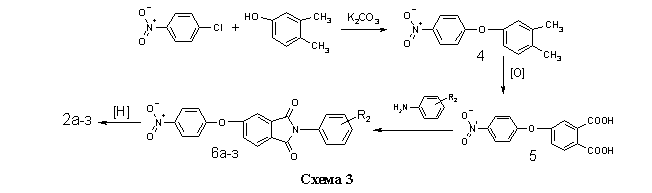
Соединения 2а-з получали на основе п-нитрохлорбензола и 3,4-ксиленола, через стадии образования 4-нитро-3/,4/диметилдифени-локсида 4, 4-нитрофеноксифталевой кислоты 5 и нитрофенокси-N-фенилфталимидов 6а-з (схема 3) по методикам, изложенным в работах [12,13].
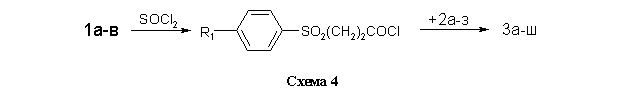
Для получения библиотеки структурных аналогов 3а-ш использованы две метода ацилирования аминов 2а-з. В первом случае (метод А, см. Экспериментальную часть) в качестве активированного ацилирующего агента использовались хлорангидриды кислот, полученных обработкой 1а-в тионилхлоридом в бензоле (схема 4).
Во втором случае (метод Б, см. Экспериментальную часть) в качестве электрофильного ацилирующего агента использовались не хлорангидриды кислот 7а-в, а их имидазолилы 8а-в, полученные взаимодействием 1а-в с N,N-карбонилдиимдазолом 9 в безводном диоксане. Ввиду малой основности 9 и слабого характера амидной связи в имидазолилах 8а-в, последние легко вступают в реакцию переамидизации с аминами 2а-з (схема 5).
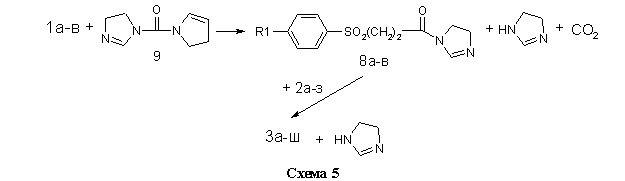
Данный метод находит в последнее время всё большее использование в органическом синтезе. Его очевидным преимуществом является отсутствие необходимости в использовании высокотоксичного тионилхлорида для получения активного ацилирующего агента и лёгкость очистки целевого продукта от побочного имидазола.
Таблица
| Соединение | R1 | R2 | Метод получения и способ очистки | Выход, % | Температура плавления, °С |
| 1а | CH3 | [10,11], кр. из этанол+вода | 80 | 113...5 | |
| 1б | Cl | - | 75 | 145...6 | |
| 1в | Br | - | 80 | 154...6 | |
| 2а | p-CH3 | [12,13 ] | 70 | 181...3 | |
| 2б | m-CH3 | - | 75 | 166...8 | |
| 2в | p-OCH3 | - | 75 | 194...5 | |
| 2г | o-OCH3 | - | 70 | 202...4 | |
| 2д | p-OC2H5 | - | 75 | 195...7 | |
| 2е | p-NHCOCH3 | - | 65 | 198...200 | |
| 2ж | o-SCH3 | - | 70 | 158...9 | |
| 2з | o-CF3 | - | 70 | 190...3 | |
| 3а | CH3 | p-CH3 | А, крист. в этаноле | 80 | 257...8 |
| 3б | - | m-CH3 | - | 80 | 187...9 |
| 3в | - | p-OCH3 | 85 | 222...4 | |
| 3г | - | o-OCH3 | А, крист. в изопропаноле | 75 | 253...6 |
| 3д | - | p-OC2H5 | Б, крист. в изопропаноле | 75 | 201...4 |
| 3е | - | p-NHCOCH3 | Б, крист. в пропанол+ДМФА | 70 | 260...2 |
| 3ж | - | o-SCH3 | Б, экстр. бензолом | 80 | 219...21 |
| 3з | - | o-CF3 | - | 85 | 197...9 |
| 3и | Cl | p-CH3 | А, крист. в этаноле | 85 | 202...4 |
| 3к | - | m-CH3 | - | 80 | 184...6 |
| 3л | - | p-OCH3 | - | 85 | 269...71 |
| 3м | - | o-OCH3 | А, крист. в диоксан+вода | 80 | 225...7 |
| 3н | - | p-OC2H5 | Б, крист. в диоксан+вода | 70 | 259...61 |
| 3о | - | p-NHCOCH3 | Б, крист. в пропанол+ДМФА | 70 | 272...5 |
| 3п | - | o-SCH3 | Б, крист. в изопропаноле | 75 | 178...80 |
| 3р | - | o-CF3 | Б, крист. в изопропаноле | 70 | 185...7 |
| 3с | Br | p-CH3 | А, экстр. диэтил. эфиром | 75 | 253...7 |
| 3т | - | m-CH3 | А, экстр. диэтил. эфиром | 70 | 212...4 |
| 3у | - | p-OCH3 | А, крист. в диоксан+вода | 65 | 265...7 |
| 3ф | - | o-OCH3 | А, крист. в диоксан+вода | 65 | 137...9 |
| 3х | - | p-OC2H5 | Б, крист. в изопропаноле | 70 | 249...50 |
| 3ц | - | p-NHCOCH3 | Б, крист. в пропанол+ДМФА | 70 | 279...82 |
| 3ч | - | o-SCH3 | Б, экстр. бензолом | 75 | 164...7 |
| 3ш | - | o-CF3 | - | 80 | 209...11 |
В общей сложности, в данной работе было синтезировано 24 не описанных ранее в литературе соединения 3а-ш, содержащих имидный и сульфонильный фрагменты. Их структура, методы получения и очистки, выход и температура плавления представлены в таблице. Выход 1а-в дан в расчёте на исходное бензольное производное (схема 2), выход 2а-з дан в расчёте на кислоту 5 (схема 3), выход 3а-ш дан в расчёте на кислоту 1а-в (схема 1).
Для первоначальной оценки биологической активности полученных соединений проведено компьютерное моделирование с помощью системы PASS, позволяющей выполнить прогноз более 500 видов биологической активности исходя из структурной формулы химического соединения и другой эмпирической информации [6,7]. Оптимальное сочетание предсказательных индексов биологической активности (Ра) и инактивности (Рi) в отношении к определённому фармакологическому эффекту получено (в рамках системы PASS) для 3г (Ра=0,512; Рi=0,064; антиконвульсант), 3ф (Ра=0,505; Рi=0,072; антагонист рецепторов GABA), 3ч (Ра=0,584; Рi=0,049; активатор калиевых каналов).
Таким образом, в результате проведённой экспериментальной работы на основе различных карбоновых кислот синтезированы библиотеки структурных аналогов гетероциклического типа, содержащих одновременно имидный и сульфонильный фрагменты. Строение и чистота синтезированных соединений подтверждалась определением температуры плавления, методом тонкослойной хроматографии, ИК- и ПМР-спектроскопией. Проведённое компьютерное моделирование позволило выявить факт влияния структурных параметров на биологическую активность полученных соединений. Ряд синтезированных соединений передан для дальнейших исследований в Институт физиологически активных веществ РАН.
Работа проводилась при поддержке предприятия "Контакт-Сервис" (Москва). Авторы выражают благодарность доценту кафедры медицинской химии МГУ С. Е. Ткаченко за ценные советы при подготовке публикации.
Экспериментальная часть
Реакцию получения 3а-ш проводили через стадию образования активных ацилирующих агентов двумя методами.
Метод А (через хлорангидриды кислот 1а-в). Синтез хлорангидридов 7а-в проводили путем кипячения исходной кислоты в тионилхлориде в присутствии каталитических количеств ДМФА в течение 1 ч с последующей отгонкой тионилхлорида. В качестве растворителя для реакции получения целевых амидов 3 использовали безводный ДМФА, в который добавляли пиридин в шестикратном мольном избытке по отношению к хлорангидриду 7а-в. Роль пиридина заключалась в связывании хлористого водорода, выделяющегося в процессе реакции. Это препятствовало протеканию побочной реакции хлористого водорода с исходным амином. Реакцию проводили при эквимолярном соотношении хлорангидрида 7 и амина 2 и температуре 10...40 оС в течение 3 ч. Охлаждённую реакционную массу выливали в воду, выпавший осадок фильтровали. Продукт сушили, кристаллизовали (или экстрагировали) и анализировали.
Метод Б (через амиды кислот 1а-в с N,N-карбонилдиимдазолом). Синтез амидов 8а-в проводили следующим образом. В стеклянную колбу, снабженную мешалкой, холодильником и термометром, загружали 1 и 9 в соотношении 1.0:1.2 (моль) и диоксан из расчёта 1 г 9 - 1 мл диоксана. Реакцию проводили при постоянном перемешивании, температуре 40...500С в течение 1 часа. Далее в колбу загружали амин 2 в соотношении 1:2=1:0,95 (моль). Амин использовали в небольшом недостатке по отношению к кислоте, исходя из того, что дальнейшая очистка от исходного амина проходит труднее, чем от исходной кислоты. Реакцию проводили при кипении диоксана в течение 1 ч. Охлаждённую реакционную массу выливали в 5%-ный раствор соды, выпавший осадок фильтровали. Продукт сушили, кристаллизовали (или экстрагировали) и анализировали.
Ход реакции 2а-з с ацилирующими агентами 1а-в контролировали методом ТСХ на пластинках Silufol 254 UV c использованием элюента петролейный эфир - бензол - ацетон - уксусная кислота - 10:5:10:0.5 (об). Для целевых амидов 3а-ш в указанном элюэнте Rf составляет 0.45...0.55.
ИК-спектры записывали на приборе UR-20. Анализируемые вещества находились в виде суспензии в вазелиновом масле, тонким слоем нанесенной на призму из хлористого или бромистого натрия. Отнесение полос поглощения проводили согласно имеющимся литературным данным [14]. Для ИК-спектров целевых амидов 3а-ш характерными являются полосы поглощения в области (см-1) 3270 (N-H), 1765, 1700 (C=O имид), 1655, 1530 (C=O амид), 1240 (С-О-С).
Спектры ЯМР 1Н 5 %-ных растворов образцов в ДМСО-d6 с внутренним стандартом ТМС записаны на приборе "Brucker-AC-400P" в ИОХ РАН (Москва). Для ЯМР спектров амидов 3а-ш характерно наличие сигналов протонов ароматических ядер (6,5-8.5 м.д) и два триплета в области 3,9 и 3,7 м.д., соответствующие двум -СН2- группам. Для протона -NH- группы характерен синглет в области 10-11 м.д.
Список литературы
Зефирова О. Н., Зефиров Н. С. Медицинская химия (Medicinal chemistry). 1. Краткий исторический очерк, определения и цели // Вестн. Моск. ун-та. Сер.2. Химия. 2001. Т. 41. С. 43-47.
Стратегия и тактика орг. синтеза // III Всерос. симп. по орг. химии. 3-6 марта 2001г. Ярославль, 2001.
Машковский М. Д. Лекарственные средства. 12-е изд., перераб. и доп. М.: Медицина, 1993. 335 с.
Джилкрист Т. Химия гетероциклических соединений: Пер. с англ. / Под. ред. М. А. Юровской. М.: Мир, 1996. 463 с.
1 Всерос. конф. по химии гетероциклов памяти А. И. Коста // 19-23 сент. 2000 г. Суздаль. М., 2000.
Филимонов Д.А., Поройков В.В. Компьютерная оценка свойств химических соединений с помощью системы PASS // Хим.-фарм. ж. 1998. Т.32. С. 32-39.
Филимонов Д. А., Поройков В. В. Средняя точность прогноза при оценке фармакологических эффектов ряда биологически активных веществ с помощью системы PASS // Вопросы мед. химии. 1997. Т. 43. С. 41-57.
Зефирова О. Н., Зефиров Н. С. Медицинская химия (Medicinal chemistry). 2. Методологические основы создания лекарственных препаратов. // Вестн. Моск. ун-та. Сер.2. Химия. Т. 41. №2. С.103-108.
Яхонтов Л. Н., Глушков Р. Г. Синтетические лекарственные средства. М.: Медицина, 1983. 273 с.
Фельдман И. Х., Михайлова В. Н. Синтез ароматических сульфопропионовых и сульфоуксусных кислот // Журн. общ. хим. 1963. №7. С.2111-2114.
Органикум.: Практикум по органической химии. Т.2: Пер. с нем. / Под ред. В. М.Потапова и С.В.Пономарева. М.: Мир, 1979. С.257.
Dorogov M. V., Nosova G. I., Koshel G. N. et al. Synthesis of multinuclear asymmetrical diamines based on 4-(nitrophenoxy)phthalic asids // Mendeleev Comm. 1997. Nr.1. Р.38-40.
Дорогов М.В., Буданов Н.А., Лебедева Н.А. и др. Окисление (нитрофенокси)-о-ксилолов до нитрофеноксифталевых кислот // Журн. прикл. химии. 1997. Т.70. Вып.10. С.1694-1697.
Наканиси К. Инфракрасные спектры и строение органических соединений: Пер с англ. М.: Мир, 1965. 191 с.
Для подготовки данной работы были использованы материалы с сайта http://www.yspu.yar.ru