Учебное пособие: Химия отрасли
Министерство образования И НАУКИ РФ
ФЕДЕРАЛЬНОЕ АГЕНСТВО ПО ОБРАЗОВАНИЮ
Иркутский государственный технический университет
ХИМИЯ ОТРАСЛИ
Лабораторный практикум
Издательство
Иркутского государственного технического университета
2007
Рецензент: канд. хим. наук, доцент ИрГТУ Е.А.Привалова
Химия отрасли. Лаб. практикум. Составитель Евстафьев С.Н. – Иркутск: Изд-во ИрГТУ. – 2007 – 65 с.
Приведены методики, применяющиеся при технохимическом контроле качества сырья, полуфабрикатов и готовой продукции пивоваренного, ликероводочного, винодельческого и безалкогольного производств.
Практикум предназначен для студентов специальности 260204 «Технология бродильных производств и виноделие» в рамках освоения дисциплины «Химия отрасли».
Печатается по решению редакционно-издательского совета Иркутского государственного технического университета
Введение
Важнейшим элементом в решении задачи выпуска продукции высокого качества является технохимический контроль производства, заключающийся в проверке исходного сырья и материалов при поступлении на производство, в период хранения и переработки, а также в оценке качества готовой продукции.
Технохимический контроль направлен на улучшение качества продукции, внедрение рациональных технологий, соблюдение норм расхода сырья и материалов, снижение их потерь.
Настоящий практикум является руководством для выполнения лабораторных работ по дисциплине «Химия отрасли», проводимых с целью закрепления теоретических знаний и приобретения практических навыков в проведении необходимых анализов по контролю производства.
Лабораторный практикум по характеру и содержанию работ подразделяется на пять разделов: контроль качества сырья, контроль качества пива, контроль качества водки, контроль качества вина и виноматериалов, контроль качества безалкогольных напитков и минеральных вод. Он охватывает основные разделы дисциплины «Химия отрасли» и включает 14 лабораторных работ.
В начале раздела или перед началом лабораторной работы дается введение, в котором поясняется сущность данного этапа контроля, а также излагаются основные теоретические положения, необходимые для выполнения работы. Приводится цель работы, необходимое оборудование и реактивы, методика проведения работы и контрольные вопросы.
Порядок выполнения лабораторных работ
Студенту необходимо заблаговременно подготовиться к выполнению работы, глубоко изучить соответствующий теоретический материал по лекциям или учебникам, а также по лабораторному практикуму, познакомиться с нормативно-технической документацией по теме.
При этом студент должен усвоить состав и свойства изучаемого объекта, сущность биохимических и физико-химических превращений в ходе технологических процессов его переработки или получения; цель работы, важность определяемых в работе показателей и их влияние на качество перерабатываемого сырья, полуфабрикатов и готовой продукции, методику проведения работы и принципы, положенные в основу определения того или иного показателя, устройство прибора или установки.
Вопросы, возникающие при самостоятельной подготовке к работе, студент должен выяснить у преподавателя, ведущего лабораторный практикум.
В начале занятия преподаватель путем опроса выясняет подготовленность студентов к работе.
Студенты, допущенные к работе, приступают к ее выполнению в соответствии с методикой, изложенной в практикуме.
Работая в лаборатории, студенты обязаны неукоснительно соблюдать правила техники безопасности, правила личной и производственной гигиены. К работе приступают, надев санитарную одежду (халат). На всех рабочих местах должен быть порядок. По окончании лабораторного занятия посуду необходимо немедленно вымыть, приборы отключить, реактивы поставить на соответствующее место.
Все данные, получаемые в ходе работы (показания приборов, расчеты и др.) заносятся в рабочую тетрадь, обрабатываются и заносятся в сводную итоговую таблицу, после анализа которых делаются соответствующие выводы.
На следующем занятии студент сдает преподавателю оформленный отчет (в рабочей тетради) по выполненной работе.
В отчете должны быть указаны цель работы, краткое описание устройства приборов или установки, подробный расчет определяемых величин, анализ полученных данных и соответствующие выводы. Каждую выполненную и оформленную работу студент защищает у преподавателя и получает зачет.
Раздел I. КОНТРОЛЬ КАЧЕСТВА СЫРЬЯ
Лабораторная работа № 1
Определение содержания крахмала в ячмене
Крахмал самая важная составная часть экстракта ячменя, определяющая его производственную и экономическую ценность. Хороший пивоваренный ячмень должен содержать 58–65 % крахмала. Обычно, чем больше в ячмене содержится крахмала, тем выше выход экстракта. Разница между содержанием крахмала в ячмене и его экстрактивностью находится в пределах 10–20 %.
Основным методом, применяемым для определения содержания крахмала, является поляриметрический метод Эверса, суть которого заключается в гидролизе крахмала до сахаров при кипячении в растворе соляной кислоты. Одновременно происходят образование декстринов и частичный переход в раствор оптически активных веществ, таких как пентозаны и белки. После осаждения белков с помощью молибдата аммония раствор сахаров поляризуют. Точность определения крахмала зависит от степени измельчения зерна.
Приборы: сахариметр; водяная баня; широкогорлая колба на 100 см3; пипетки на 1 и 25 см3; весы технические; лабораторная мельница.
Реактивы: 1,124 % –ный раствор соляной кислоты: 24,9 см3 соляной кислоты относительной плотности 1,19 разбавляют дистиллированной водой до 1 дм3; 14,5 %-ный раствор молибдата аммония.
Ход анализа. Навеску 5 г тонко измельченного зерна (помол должен проходить через сито с отверстиями 0,5 мм; на сите может оставаться лишь небольшое количество шелухи) помещают в широкогорлую мерную колбу емкостью 100 см3 с 25 см3 раствора 1,124 %-ной соляной кислоты. Перемешиванием достигают полного смачивания муки, комков допускать нельзя. Доливают еще 25 см3 того же раствора соляной кислоты, смывая им частицы муки со стенок горла колбы. Колбу помещают на кипящую водяную баню, причем первые 3 минуты содержимое колбы размешивают круговым движением. Колба должна быть погружена в баню до самого горлышка.
Через 20 минут нагревания из содержимого колбы берут пробу на анализ содержания крахмала (йодная проба). В случае положительного результата нагревание продолжают в течение 15 минут. После чего, вынув колбу из водяной бани, вливают в нее 35–40 см3 холодной дистиллированной воды и содержимое колбы охлаждают до 20 °С, затем добавляют 2 см3 14,5 %-ного раствора молибденовокислого аммония, колбу доливают до метки водой, энергично взбалтывают и фильтруют через складчатый фильтр в сухую колбу, первые порции фильтрата перефильтровывают.
Поляризацию надо производить немедленно. Отсчет делают не менее 3–х раз, для вычислений берут среднее арифметическое.
Содержание крахмала (С, в % на воздушно-сухое вещество) рассчитывают по формуле:
|
V . α . 100 C = –––––––––– , [α]D . l . S |
где: V – объем колбы, взятой для анализа (100 см3);
α – угол вращения, град.;
[α]D – удельное вращение испытуемого вещества (181,5 для ячменя);
l – длина поляризационной трубки (0,94 дм);
S – навеска ячменя, г.
При использовании поляриметра с линейной шкалой формула приобретает следующий вид:
|
V . α . 100 C = ––––––––– . 0,3468 , [α]D . l . S |
где: 0,3468 – коэффициент перехода от линейной шкалы поляриметра к круговой.
Содержание крахмала (С1, в % на сухое вещество) рассчитывают по формуле:
|
С . 100 С1 = ––––––– , 100 – w |
где: С – содержание крахмала на воздушно-сухое вещество, %;
w – влажность зерна, %.
Контрольные вопросы:
1. Крахмал. Его строение, состав и свойства.
2. Кислотный гидролиз крахмала. Недостатки метода.
3. Крахмалистость ячменя, значения этого показателя для производства солода и пива.
4. Принципы поляриметрического метода определения крахмалистости ячменя.
Лабораторная работа № 2
Определение пленчатости зерна
Пленчатостью называют количество мякинной оболочки, выраженное в процентах от общей массы зерна.
Толщина (количество) мякинной оболочки ячменя, кроме чисто химического влияния на состав полученного сусла и пива, оказывает влияние на рентабельность переработки того или другого сорта ячменя. Чем меньше оболочка, тем больше выхода экстракта можно ожидать от ячменя. Тонкокожие ячмени содержат 6 –7 % мякинной оболочки, толстокожие – 10 % и выше. Кроме того, толстая пленка содержит больше дубильных и горьких веществ, понижающих качество пива.
Мякинная оболочка ячменя прочно приклеена к ядру пектиновыми веществами и, чтобы снять их, нужно сначала растворить эти вещества. Для определения пленчатости пользуются методами Омарова (обработка горячей щелочью) и Люффа (обработка слабым раствором аммиака).
Метод Люфа
Приборы и реактивы: конические колбы на 100–150 см3; водяная баня; стеклянная или фарфоровая чашка; 5 %-ный раствор аммиака.
Ход анализа. Взвешивают на аналитических весах 50 зерен ячменя, переносят их в толстостенную склянку емкостью 100–150 см3, снабженную хорошо притертой стеклянной или плотной резиновой пробкой, приливают 10см3 5%-ного раствора аммиака, завязывают пробку тонкой проволокой или шпагатом и помещают склянку в водяную баню, нагретую до 80 °С, так глубоко, чтобы вода снаружи склянки стояла несколько выше внутреннего уровня. Температуру 80 °С поддерживают 1 час, после чего сливают аммиачный раствор с зерен.
Зерна высыпают в стеклянную или фарфоровую чашку и осторожно сдирают с них оболочку, сначала прилегающую к зародышу спинную часть, а затем брюшную. Оболочку высушивают при 105 °С в течение 3 часов и взвешивают. При пересчете необходимо иметь в виду, что во время обработки аммиаком оболочка теряет в среднем 1/12 часть своего веса.
Расчет пленчатости в % на воздушно–сухое вещество ведут по формуле:
|
|
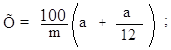
где: а – масса сухих пленок, г;
m – масса 50 зерен ячменя.
Расчет пленчатости в % на сухое вещество ведут по формуле:
|
|
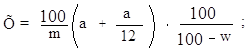
где: w – влажность ячменя, %.
Контрольные вопросы:
1. Строение зерна ячменя;
2. Оболочка зерна, ее состав;
3. Влияние пленчатости на качество ячменя и пива.
Лабораторная работа № 3
Определение содержания пентозанов в зерне
Пентозаны относятся к высшим углеводам, которые при гидролизе дают пентозы (арабинозу, ксилозу). В ячмене содержание пентозанов колеблется от 7 до 12 %. Значение пентозанов в технологии пивоварения очень большое – из этого резерва черпаются вещества для повышения выходов экстракта.
Определение основано на образовании фурфурола при действии соляной кислоты на пентозаны (метод Толленса).
Оборудование: установка для атмосферной перегонки; двугорловая круглодонная колба на 300–500 см3; цилиндр, мерная коническая колба на 500 см3, делительная воронка.
Реактивы: 12 %-ный раствор соляной кислоты, смесь анилина и уксусной кислоты (3 : 2), флороглюцин, дистиллированная вода.
Ход анализа: Собирают установку для атмосферной перегонки. В качестве перегонной колбы используют двугорловую круглодонную колбу, к которую вставлена делительная воронка с двумя метками 30 и 60 см3. В качестве приемника служит мерный цилиндр с метками 30 и 60 см3.
В перегонную колбу отвешивают 2,8–3,2 г размолотого ячменя, приливают 100 см3 12 %-него раствора соляной кислоты и нагревают колбу до 140–150°С. Нагревание обычно рекомендуется вести в чашках со сплавом Вуда или Розе, но можно вести его просто на асбестовой сетке при кипении жидкости с таким расчетом, чтобы в течение 10–15 минут в приемнике скопилось 30 см3 дистиллята.
Как только в приемнике накопится 30 см3 дистиллята, приемник освобождают, сливая дистиллят в приемную колбу с меткой 400 см3, и закрывают ее пробкой. В перегонную колбу через делительную воронку приливают порцию соляной кислоты в количестве 30 см3. Повторяют операцию, примерно, десять раз и прекращают дистилляцию, когда капля дистиллята перестанет окрашивать в малиновый цвет кусочек фильтровальной бумаги, смоченной смесью анилина и уксусной кислоты, взятых в пропорции 3:2.
К дистилляту прибавляют растворенный в нескольких миллилитрах 12 %-ного раствора соляной кислоты флороглюцин, которого берут двойное количество (по весу) от предполагаемого по расчету количества фурфурола (0,3г). Содержимое приемной колбы доливают 12 %-ным раствором соляной кислоты до метки 400 и хорошо размешивают. Через 3 часа проверяют полноту осаждения по реакции с уксуснокислым анилином; окрашивания не должно быть, в противном случае добавляют еще некоторое количество флороглюцина, через 3 часа вновь определяют полноту осаждения и оставляют отстаиваться в течение ночи.
Выпавший зеленовато-черный осадок собирают на высушенном и взвешенном фильтре, промывают, примерно, 150 см3 дистиллированной воды (операцию фильтрования и промывания можно вести при некотором разрежении). Отфильтрованный осадок, поместив его с фильтром в бюкс, высушивают при 98–100 °С в течение 3,5 – 4 часов и после охлаждения в эксикаторе взвешивают.
По полученной массе осадка, пользуясь диаграммой Приложения 1, находят количество пентозанов во взятой навеске и пересчитывают его на 100 г сухого вещества.
Контрольные вопросы:
1. Дайте характеристику пентозанам: строение, состав, свойства.
2. Какие моносахариды получаются при гидролизе пентозанов?
3. Напишите уравнения химических реакций превращения пентозанов в фурфурол.
Лабораторная работа № 4
Определение аминного азота в мелассе
Аминный азот представлен в растительном сырье аминокислотами, пептидами и белками, которые являются источником азотного питания дрожжей, и содержание их в процессе брожения заметно снижается. В результате их превращений под действием дрожжей образуются высшие спирты. При термической обработке аминокислоты и пептиды, вступая во взаимодействие с сахарами, образуют меланоидины, альдегиды и другие продукты, оказывающие существенное влияние на качество конечной продукции.
Химизм процесса при определении аминного азота медным способом сводится к следующему. При взаимодействии натриевой соли аминокислоты с суспензией фосфата меди образуется окрашенная в синий цвет хорошо растворимая комплексная медная соль аминокислоты:
|
|
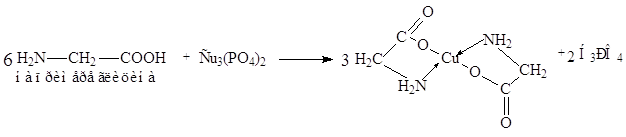
Фосфорная кислота связывается боратным буфером, и реакция идет до конца.
В фильтрате после отделения избытка фосфата меди оказываются лишь медные соли аминокислот (за исключением цистина, медная соль которого нерастворима), и, следовательно, по количеству меди, перешедшей в фильтрат, можно определить содержание аминокислот.
При добавлении к фильтрату концентрированной уксусной кислоты, последняя вытесняет из медной соли более слабую аминокислоту:
![]() (H2N - СН2 - СОО)2Сu + 2СН3СООН 2H2N - СН2 - СООН + (СН3СОО)2 Сu
(H2N - СН2 - СОО)2Сu + 2СН3СООН 2H2N - СН2 - СООН + (СН3СОО)2 Сu
Под действием йодоводородной кислоты, образовавшейся из йодида калия в кислой среде, ион меди со степенью окисления +2 восстанавливается, образуется нерастворимый йодид меди (I) и свободный йод:
![]()
![]() 2(СН3СОО)2Сu + 4 HI 2CuI +
4СН3СООН + 12
2(СН3СОО)2Сu + 4 HI 2CuI +
4СН3СООН + 12
Вследствие нерастворимости йодида меди (I) в слабокислой среде этот процесс также идет до конца. Таким образом, количество выделившегося свободного йода эквивалентно количеству медных солей аминокислот. Концентрацию свободного йода определяют титрованием выделившегося йода раствором гипосульфита:
![]() 2Na2S2O3 + I2 2NaI + Na2S4O6
2Na2S2O3 + I2 2NaI + Na2S4O6
гипосульфит тетратионат натрия
По уравнению реакции 0,5 моль выделившегося йода соответствует 1 мо–ль меди, который в свою очередь эквивалентен 28 г аминного азота. С другой стороны, 0,5 моль йода реагирует с одним грамм-эквивалентом гипосульфита. Следовательно, один грамм-эквивалент гипосульфита соответствует 28 г аминного азота. Отсюда 1 см3 0,01н раствора гипосульфита отвечает 0,28 мг аминного азота. Умножением величины 0,28 мг на затраченный объем 0,01н раствора гипосульфита (минус контроль) получают количество миллиграммов аминного азота во взятом, объеме (10 см3) испытуемого раствора. После этого делают пересчет на весь объем раствора в колбе и сравнивают найденное количество аминного азота с тем, что должно быть в 2 см3 исследуемого раствора мелассы:
|
0,28 . V . 12.5 С = –––––––––––– , m |
где: V – объем гипосульфита, пошедший на титрование;
12,5 – коэффициент разбавления;
m – масса мелассы на 100 г раствора.
Оборудование: фильтры бумажные, колбы мерные на 25 см3 (2 шт.), пипетки с меткой на 1, 2 и 10 см3, бюретки прямые с краном на 25 или 50 см3, воронки для фильтрования, колбы конические на 100 см3 (4 шт.), цилиндр мерный с носиком на 10 см3.
Реактивы: раствор хлорида меди (II): 27,3 г в 1 дм3 раствора; раствор фосфата натрия: 68,5 г Na3PO4 ·12H2O в 1 дм3 раствора или 64,5 г Na2HPO4·12H2O растворяют в 500 см3 дистиллированной воды, из которой кипячением удален СО2, и добавляют 7,2 г NaOH с последующим доведением объема раствора до 1 дм3 дистиллированной водой; боратный буферный раствор; 28,6 г тетрабората натрия растворяют в 750 см3 воды, добавляют 50см3 1н раствора соляной кислоты и доводят водой до 1 дм3; рН=8,8; суспензия фосфата меди: смешивают один объем раствора хлорида меди (II) с двумя объемами раствора фосфата натрия и приливают два объема боратного буфера, суспензию готовят только перед работой в необходимом объеме; тимолфталеин: 0,25 %-ный в этиловом спирте (50 %-ом); 0,1н раствор Na2S2O3·5H2O (из этого раствора разбавлением готовится 0,01 н раствор, титр которого устанавливают по точному раствору 0,01 н йода калия); крахмал (1 %-ный), йодид калия (10 %-ный), уксусная кислота (конц.), гидроксид натрия (0,5 н), меласса (13 %-ный раствор).
Ход анализа. В мерную колбу на 25 см3 берут 2 см3 исследуемого раствора мелассы, добавляют 2 капли тимолфталеина и по каплям 0,5н раствор гидроксида натрия до слабоголубого окрашивания (рН раствора 10,2). После этого добавляют 10 см3 суспензии фосфата меди, хорошо перемешивают. При исчезновении осадка следует добавить еще 5 см3 суспензии. Раствор в колбе доводят до метки водой, тщательно перемешивают многократным переворачиванием колбы и отфильтровывают избыток фосфата меди через плотный фильтр. Фильтрат должен быть совершенно прозрачным. Этого добиваются многократным фильтрованием. Из фильтрата пипеткой берут две пробы по 10см3 в конические колбы для титрования, подкисляют 0,4 см3 концентрированной уксусной кислоты, добавляют 6–8 см3 10%-ного раствора йодида калия и выделившийся йод титруют 0,01н раствором гипосульфита. Крахмал в количестве 1–2 см3 (20–40 капель) на 100 см3 раствора добавляют в тот момент, когда титруемый раствор примет соломенно-желтую окраску. Титрование продолжают до исчезновения появившейся после добавления раствора крахмала синей окраски.
Ставят контрольное определение, в котором вместо мелассы берут такой же объем воды. Количество гипосульфита, затрачиваемое на контрольный раствор, вычитают из такового в опыте.
Контрольные вопросы:
1. Белки. Их строение состав и свойства;
2. Содержание азотистых веществ в ячмене. Их роль в пивоварении;
3. Химизм процесса определения аммиачного азота медным способом.
Раздел 2. КОНТРОЛЬ КАЧЕСТВА ПИВА
Завершающим этапом технологического контроля производства пива является оценка качества готового продукта, которая осуществляется по органолептическим и физико–химическим показателям.
Главнейшими показателями качества пива как напитка являются прозрачность, цвет, вкус, аромат, хмелевая горечь, пенообразование. Все эти свойства пива определяются в процессе дегустации.
К физико–химическим показателям пива относят массовую долю сухих веществ в начальном сусле, массовую долю спирта и действительного экстракта, кислотность, цветность, массовую долю двуокиси углерода (для бутылочного пива), стойкость пива, время дображивания.
Лабораторная работа № 5
Определение горьких веществ в пиве
Вкус пива относят к органолептическим показателям. Горький вкус пива вызван в основном горькими веществами хмеля, а также дубильными веществами как хмеля, так и оболочки солода и несоложенных злаков. Горькие вещества хмеля представлены мягкими и твердыми смолами. Мягкие смолы – это α–кислота (гумулон), β–кислота (лупулон), мягкие α– и β–смолы, γ–кислота (гумулион), σ–кислота (гулупон), а также β–фракция (сумма β–кислот и мягких смол). Твердые смолы разделяют на γ– и σ–смолы.
Среди горьких веществ наибольшая доля приходится на α–кислоту. При сушке и хранении хмеля она окисляется и полимеризуется с образованием мягкой α–смолы, а при более глубоком окислении – твердой смолы. При кипячении сусла α–кислота превращается в изо–α–кислоту, обладающую большей горечью и растворимостью в воде.
В составе β–кислот преобладает лупулон, который при кипячении сусла остается в хмелевой дробине. При окислении превращается в β–смолы, растворимые в сусле и пиве.
|
|
|
|
α-кислоты |
изо–α-кислота |
|
R: изобутил (гумулон); изопропил (когумулон); втор–бутил (адгумулон) |
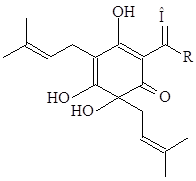
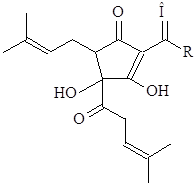
Растворимость мягких смол в сусле выше, чем кислот, из которых они образованы.
Твердые смолы, образующиеся при окислении кислот, обладают незначительной горечью, только σ–твердая смола имеет грубую горечь и хорошо растворима в сусле и пиве.
В процессе получения пива часть горьких веществ теряется из–за их адсорбции на дрожжах и при всплытии на пузырьках СО2. Лишь 20–40 % исходных горьких веществ присутствует в пиве.
Растворителем горьких веществ в пиве также, как и в сусле, служит хлороформ, причем надо отметить, что из всего комплекса горьких веществ α-горькая кислота (гумулон) при рН пива обладает наибольшей растворимостью и во много раз превосходит в этом отношении β-горькую кислоту (лупулон).
Оборудование: колбы на 75, 100 и 1000 см3, делительные воронки на 100 и 1000 см3, центрифуга, пипетка, бумажный фильтр, эксикатор, сушильный шкаф.
Реактивы: раствор серной кислоты, хлороформ, безводный сернокислый натрий.
Ход анализа: Пиво предварительно освобождают от главной массы углекислоты взбалтыванием и фильтрованием. Для чего пиво в количестве 250-400 см3 наливают в колбу емкостью около 1 дм3 и при комнатной температуре взбалтывают его, закрыв горло сосуда ладонью и приоткрывая сосуд, время от времени, пока прекратиться ощущение давления изнутри. Непрозрачное пиво следует отфильтровать.
400 см3 (V) пива помещают в колбу со стеклянной притертой пробкой емкостью 750 – 1000 см3, подкисляют их 5 см3 разведенной серной кислоты и добавляют 50 см3 хлороформа. Колбу устанавливают в «трясучку» и содержимое подвергают энергичному встряхиванию в течение 30 минут, после чего переливают его в делительную воронку емкостью 500—750 см3 и дают в течение нескольких часов отслоиться водной части от хлороформенной. Образовавшуюся хлороформенную эмульсию для разрушения подвергают центрифугированию в течение 10 – 15 минут при 2000 – 3000 об/мин. Водную часть в центрифужных пробирках тщательно декантируют, а прозрачный хлороформенный экстракт, отстоявшейся на дне пробирок, отсасывают пипеткой, просовывая копчик ее через слой светло-серой массы, скопившейся над хлороформом, и собирают его в делительную воронку на 80 – 100 см3.
После 5–10-минутного отстаивания хлороформенный слой собирают в колбочку емкостью 100 – 150 см3 с притертой стеклянной пробкой. В колбочку предварительно помещают около 15 г безводного сернокислого натра для сушки хлороформенного экстракта. Содержимое колбочки в течение 5 – 10 минут перемешивают, после чего хлороформенный экстракт фильтруют через небольшой бумажный фильтр.
30 см3 (V1) фильтрата отбирают пипеткой в предварительно взвешенную круглодонную колбочку емкостью около 75 см3. Хлороформ отгоняют под вакуумом на кипящей водяной бане до полного его удаления. Колбочку с остатком выдерживают 1 час в эксикаторе (лучше в вакуум-эксикаторе с водоотнимающим веществом) и взвешивают. Допускается отгонка хлороформа без вакуума на водяной бане при 65–70 °С, а остаток в круглодонной колбе можно подвергать сушке в сушильном шкафу при 70 °С в течение 1 часа. Содержание горьких веществ (Х) в граммах в 1 дм3 пива находится по формуле:
|
|
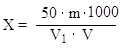
где: m – вес остатка в колбе после отгонки хлороформа, в г;
V – объем пива, взятого для анализа, в см3;
V1– объем фильтрата для анализа, в см3
Нормально в пиве содержится около 0,03-0,09 г горьких веществ в 1 дм3.
Контрольные вопросы:
1. Чем обусловлен горьких вкус пива?
2. Горькие вещества хмеля и их превращения в процессе получения пива;
3. Перечислите свойства α–кислот;
4. Сущность метода определения содержания α–кислот.
Лабораторная работа № 6
Определение спирта, действительного экстракта и
расчет сухих веществ в начальном сусле
Содержание массовой доли спирта и действительного экстракта находят дистилляционным или рефрактометрическим методами. Дистилляционный метод основан на отгонке спирта из навески пива и определении относительной плотности дистиллята и остатка пива после отгонки.
Оборудование: весы аналитические с наибольшим пределом взвешивания 200 г; баня водяная; шкаф сушильный; пикнометр, установка для атмосферной перегонки.
Реактивы: калия бихромат; кислота серная; спирт этиловый ректификованный; хромовая смесь: 1,2 г бихромата калия растворяют в 100 см3 серной кислоты.
Освобождение пива от двуокиси углерода. 250-300 см3 пива наливают в колбу вместимостью 1000 см3, доводят температуру до 20 °С, затем встряхивают, закрыв колбу ладонью, периодически приоткрывая ее, до тех пор, пока прекратится ощущение давления изнутри. Встряхивание повторяют два-три раза с интервалом в 5 мин. Непрозрачное пиво фильтруют через бумажный фильтр.
6.1. Определение массовой доли спирта
В сухую круглодонную колбу взвешивают 100 г пива, предварительно освобожденного от двуокиси углерода. Добавляют 50 см3 дистиллированной воды.
Затем колбу соединяют с холодильником и отгоняют 70–80 см3 пива в предварительно взвешенную приемную колбу, установленную в сосуд с холодной водой. В приемную колбу предварительно наливают 5–10 см3 дистиллированной воды.
После отгонки к содержимому приемной колбы добавляют дистиллированной воды до массы жидкости 100 г (равной массе взятого пива) и перемешивают.
Определение относительной плотности: Пикнометр, тщательно вымытый хромовой смесью и дистиллированной водой и высушенный до постоянной массы, взвешивают с точностью до четвертого десятичного знака. Его наполняют бидистиллятом с температурой 19-21 °С немного выше метки, закрывают пробкой и погружают водяную баню с температурой (20,0±0,2) °С так, чтобы уровень воды в бане был немного выше уровня воды в пикнометре. Через 20-30 мин, не вынимая пикнометр из бани, при температуре 20 °С устанавливают уровень воды в нем так, чтобы нижний край мениска находился на одном уровне с меткой. Избыток воды в пикнометре отбирают полоской фильтровальной бумаги. Внутреннюю поверхность горловины пикнометра выше метки тщательно вытирают фильтровальной бумагой.
Пикнометр закрывают пробкой, вынимают из бани, досуха вытирают сухим полотенцем и взвешивают с точностью до четвертого десятичного знака.
Наполнение пикнометра водой, установку уровня воды в нем и взвешивание проводят 3-4 раза, пока разница по массе будет не более 0,0030 г. Вычисляют среднее арифметическое значение массы пикнометра с водой.
Затем пикнометр освобождают от воды, ополаскивают 2-3 раза испытуемым раствором (раствор дистиллята) и заполняют им пикнометр чуть выше метки.
Термостатирование, установку уровня раствора и взвешивание пикнометра проводят описанным выше способом при температуре 20 °С. Проводят не менее двух параллельных определений.
Относительную плотность раствора дистиллята (d) вычисляют по формуле:
|
|
где m – масса пикнометра с раствором дистиллята, г;
m1– масса пикнометра, г;
m2– масса пикнометра с дистиллированной водой, г.
Массовую долю спирта в процентах в зависимости от относительной плотности раствора дистиллята определяют по табл. 1 Приложения 2.
Если масса дистиллята отличается от массы пробы пива, значение, найденное по табл.1, умножают на поправочный коэффициент (К), вычисленный по формуле:
|
|
где m4 – масса дистиллята, г;
m5 – масса пива, г.
6.2. Определение массовой доли действительного экстракта
Остаток после отгонки спирта доводят в колбе дистиллированной водой до массы 100 г, перемешивают и определяют плотность пикнометром при температуре (20,0 ± 0,2) °С.
Относительную плотность раствора остатка после отгонки спирта (d1) вычисляют по формуле:
|
|
где m3 – масса пикнометра с раствором остатка после отгонки спирта, г.
Массовую долю действительного экстракта в процентах в зависимости от относительной плотности раствора остатка после отгонки спирта определяют по табл. 2 Приложения 2.
Если масса разбавленного остатка отличается от первоначальной массы пробы пива, значение, найденное по табл. 2, умножают на поправочный коэффициент (К1), вычисленный по формуле:
|
|
где m6 – масса разбавленного остатка после отгонки спирта, г.
Вычисление проводят до второго десятичного знака. Расхождение между результатами двух параллельных определений одной и той же пробы пива при доверительной вероятности Р = 0,95 по абсолютной величине не должно превышать в процентах:
0,06 — для массовой доли спирта,
0,03 — для массовой доли действительного экстракта.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений и выражают целым числом с одним десятичным знаком.
6.3. Расчет сухих веществ в начальном сусле
Массовую долю сухих веществ в начальном сусле (m9) в процентах вычисляют по формуле:
|
|
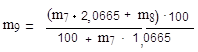
где m7 –массовая доля спирта в пиве, %;
m8 –массовая доля действительного экстракта в пиве, %;
2,0665 – масса экстракта, расходуемая на получение 1 г спирта, г;
1,0665 – масса веществ, удаляющихся при брожении с получением 1г спирта, г.
Вычисление проводят до второго десятичного знака с последующим округлением до первого десятичного знака.
Расхождение между результатами двух определений одной и той же пробы в разных лабораториях при доверительной вероятности Р = 0,95 по абсолютной величине не должно превышать 0,3 %.
Контрольные вопросы:
1. По каким показателям оценивается качество пива?
2. Какие показатели качества пива определяются физико–химическими методами анализа?
3. Какие методы используются для определения содержания спирта и действительного экстракта?
4. На чем основан дистилляционный метод определения содержания спирта?
Раздел 3. КОНТРОЛЬ КАЧЕСТВА ВОДКИ
Оценка качества водки осуществляется по органолептическим и физико–химическим показателям.
Органолептический анализ включает оценку цвета, аромата и вкуса водки. К физико–химическим показателям относят крепость, щелочность, массовые концентрации альдегидов, сивушного масла, сложных эфиров и объемную долю метанола (см. Приложение 4).
Лабораторная работа № 7
ОПРЕДЕЛЕНИЕ ФИЗИКО–ХИМИЧЕСКИХ ПОКАЗАТЕЛЕЙ ВОДОК
7.1. Определение крепости
Метод основан на измерении объемной доли этилового спирта ареометром для спирта в дистилляте, полученном после предварительной перегонки спирта из анализируемого изделия. Диапазон измерения объемной доли этилового спирта 0-100%. Метод может быть использован для определения содержания спирта в пиве, вине, водке и ликероводочных изделиях.
Оборудование: установка для атмосферной перегонки, ареометр.
Проведение анализа. 250 см3 водки, отмеренной мерной колбой соответствующей вместимости при температуре 20 °С, помещают в перегонную колбу вместимостью 500 см3. Мерную колбу ополаскивают два-три раза дистиллированной водой, сливая ее содержимое в перегонную колбу с таким расчетом, чтобы объем дистиллированной воды не превышал 60-100 см3.
Приемником для сбора дистиллята используют ту же мерную колбу, которой отмеривали анализируемую водку. В приемник наливают 10-15 см3 дистиллированной воды и погружают узкий конец стеклянной трубки холодильника для получения водяного затвора. Затем приемную колбу помещают в баню с холодной водой или льдом и начинают перегонку.
После заполнения приемной колбы примерно наполовину ее опускают так, чтобы конец трубки холодильника не погружался в дистиллят. Конец трубки холодильника ополаскивают 5 см3 дистиллированной воды и продолжают перегонку без водного затвора.
После заполнения приемной колбы дистиллятом на 4/5 объема перегонку прекращают. Колбу с дистиллятом доливают дистиллированной водой немного ниже метки и выдерживают в течении 20-30 мин при температуре 20 °С в водяной бане. Затем содержимое приемника доводят до метки дистиллированной водой и тщательно перемешивают.
Для измерения концентрации этилового спирта дистиллят наливают в стеклянный цилиндр осторожно по стенке цилиндра во избежание появления пузырьков воздуха. Цилиндр с дистиллятом помещают в водяную баню температурой 20 °С, в цилиндр опускают термометр и ареометр и выдерживают в течение 10 мин, не допуская изменений температуры. При этом ареометр берут за верхний конец стержня, свободный от шкалы, опускают в дистиллят, погружая его до тех пор, пока до предполагаемой отметки ареометрической шкалы не останется 3–4 мм, затем оставляют ареометр в покое. По истечении 3 мин снимают отсчет показаний ареометра. Ареометр должен плавать в дистилляте, не касаясь стенок цилиндра.
Отсчет показаний ареометра проводят по нижнему краю мениска с точностью до 0,2 наименьшего деления.
Дистиллят не выливать!! Он будет использован при выполнении анализа по п.п. 7.2 – 7.6.
7.2. Определение щелочности
Щелочность водки определяют химическим ацидиметрическим методом и титриметрическим методом с применением потенциометра.
Ацидиметрический метод основан на титровании определенного объема водки раствором соляной кислоты до получения нейтральной реакции, устанавливаемой по индикатору.
Реактивы: раствор соляной кислоты концентрации 0,1 моль/дм3; индикатор.
Проведение анализа. Испытуемую водку в количестве 100 см3 помещают в коническую колбу вместимостью 250 см3 и титруют ее в присутствии раствора метилового красного раствором соляной кислоты концентрации 0,1моль/дм3 до перехода желтого оттенка раствора в розовый. По объему раствора соляной кислоты, пошедшего на титрование, судят о щелочности водки.
За окончательный результат анализа принимают среднее арифметическое результатов двух параллельных определений, которое округляют до первого десятичного знака.
7.3. Определение массовой концентрации альдегидов
Массовую концентрацию альдегидов в водке определяют с помощью типовых спиртовых растворов или фотоэлектроколориметрическим методом.
Фотоэлектроколориметрический метод основан на измерении оптической плотности испытуемого раствора после реакции присутствующих в анализируемой водке альдегидов с пирогаллолом А в сернокислой среде.
Реактивы: концентрированная серная кислота; 0,1 %-ный раствор пирогаллола А.
Оборудование: фотоэлектроколориметр КФК-3.
Проведение анализа. Водки, содержащие сахар, и водку «Посольская» перед анализом предварительно подвергают перегонке по методике, изложенной в п.7.1. Полученный дистиллят используют для определения массовой концентрации альдегидов.
В пробирку с пришлифованной пробкой вносят 2 см3 концентрированной серной кислоты, затем осторожно по стенке пробирки приливают 5 см3 анализируемой водки или ее дистиллята и 1,5 см3 0,1%-ного раствора пирогаллола А, не допуская смешивания этих растворов. Пробирку закрывают пробкой, ее содержимое энергично перемешивают и выдерживают в кипящей водяной бане в течение 5 мин. Затем пробирку помещают в проточную холодную воду или в баню со льдом и охлаждают до комнатной температуры.
Образовавшуюся в результате проведенной реакции окраску раствора измеряют на фотоэлектроколориметре в кюветах с толщиной поглощающего свет слоя 10 мм при светофильтрах с длиной световой волны 440 нм по сравнению с дистиллированной водой.
Полученную величину оптической плотности используют для расчета количества альдегидов (мг/дм3 безводного спирта) по градуировочному графику, построенному по стандартным растворам ацетальдегида или по расчетному уравнению, выведенному на основании градуировочного графика (см. Приложение 3).
Расчетное уравнение имеет вид:
Сал = AD – A1;
где Сал – массовая концентрация альдегидов, мг/дм3 безводного спирта;
А и А1 — расчетные коэффициенты, полученные экспериментально;
D — оптическая плотность.
Расчетные коэффициенты в формуле необходимо определять для каждой марки фотоэлектроколориметра и партии используемого пирогаллола А. Для этого следует построить градуировочный график зависимости оптической плотности от содержания альдегидов в водке.
Построение градуировочного графика. Для построения градуировочного графика используют типовые спиртовые растворы (стандартные образцы) с содержанием альдегидов (ацетальдегида) 2; 3; 4 и 10 мг в 1 дм3 безводного спирта.
Проводят колориметрическую реакцию этих растворов с раствором пирогаллола А по вышеописанной методике.
Полученные после колориметрирования значения оптических плотностей откладывают на оси ординат, а соответствующую этим значениям массовую концентрацию альдегидов – на оси абсцисс.
Оптическую плотность каждого раствора определяют не менее трех раз и из полученных значений находят среднее арифметическое.
Зависимость между оптической плотностью и количеством альдегидов в анализируемых растворах на градуировочном графике должна быть прямолинейной.
7.4. Определение массовой концентрации сивушного масла
Сивушное масло представляет собой смесь н–пропилового, изобутилового и амиловых спиртов. По внешнему виду оно представляет собой прозрачную жидкость без механических примесей от светло–желтого до красно–бурого цвета.
Массовую концентрацию сивушного масла в водке определяют визуально с помощью типовых спиртовых растворов (стандартных эталонов) или фотоэлектроколориметрическим методом, который основан на измерении оптической плотности анализируемого раствора, полученного после реакции присутствующих в водке высших спиртов (сивушного масла) с салициловым альдегидом в присутствии серной кислоты.
Реактивы: концентрированная серная кислота; 1,0 %-ный раствор салицилового альдегида.
Оборудование: фотоэлектроколориметр КФК-3.
Проведение анализа. Концентрированную серную кислоту в объеме 10см3 вносят в пробирку с пришлифованной пробкой, осторожно по стенке пробирки приливают 5 см3 испытуемой водки с таким расчетом, чтобы не произошло смешивания обеих жидкостей, а образовалось два слоя. Затем приливают 0,7 см3 1 %-ного спиртового раствора салицилового альдегида, пробирку закрывают пробкой, содержимое энергично перемешивают и выдерживают в кипящей водяной бане в течение 10 мин, отмечая время по секундомеру. Затем пробирку погружают в проточную холодную воду или водяную баню со льдом для быстрого охлаждения реакционной смеси до комнатной температуры. Интенсивность образовавшейся в результате проведенной реакции желтой окраски измеряют не позднее чем через 5 мин на фотоэлектроколориметре любой марки при светофильтре с длиной световой волны 540 нм в кюветах с толщиной поглощающего свет слоя 20 мм, а затем сравнивают с дистиллированной водой.
Обработка результатов. Для расчета массовой концентрации сивушного масла в водке следует внести поправку на присутствующие в нем альдегиды, также реагирующие с салициловым альдегидом. Для этого из полученного после колориметрирования значения оптической плотности следует вычесть значение расчетной оптической плотности, соответствующее тому количеству альдегидов, которое определено в анализируемой водке по графику или вычислено по уравнению (см. п. 7.3). Эти значения расчетных оптических плотностей приведены в таблице:
Расчетные значения оптической плотности для определения
поправки на содержание сивушного масла в водке
|
Массовая концентрация альдегидов в водке в пересчете на уксусный, мг/дм3 безводного спирта |
Расчетные значения оптической плотности по фотоэлектроколориметрам КФК-2 и КФК-3 |
|
1,5 2,0 2,5 3,0 4,0 5,0 6,0 7,0 7,5 8,0 8,5 9,0 10,0 |
0,010 0,015 0,020 0,025 0,040 0,050 0,060 0,075 0,080 0,085 0,090 0,100 0,110 |
Величину оптической плотности, полученную после вычитания расчетной оптической плотности (поправка на альдегиды), используют для расчета количества сивушного масла (мг/дм3 безводного спирта) по градуировочному графику, построенному по стандартным растворам сивушного масла или по расчетному уравнению, выведенному на основании градуировочного графика.
Расчетное уравнение имеет вид:
Сс.м. = КD – K1,
где Сс.м. – массовая концентрация сивушного масла, мг/дм3 безводного спирта;
К и К1 — расчетные коэффициенты, полученные экспериментально;
D — оптическая плотность.
Расчетные коэффициенты в формуле необходимо определять для каждой марки фотоэлектроколориметра, используемого в работе. Для этого следует построить градуировочный график зависимости оптической плотности от содержания сивушного масла.
Построение градуировочного графика. Для построения градуировочного графика используют стандартные растворы, содержащие сивушное масло (смесь изоамилового и изобутилового спиртов (3:1)) в пересчете на безводный спирт в следующем количестве: 2; 3; 4; 8; 10 и 15 мг в 1 дм3.
Проводят колориметрическую реакцию указанных стандартных растворов с 1%-ным спиртовым раствором салицилового альдегида по методике, приведенной выше. По полученным после колориметрирования величинам оптических плотностей и массовой концентрации сивушного масла в анализируемых стандартных растворах строят градуировочный график, откладывая на оси абсцисс массовую концентрацию сивушного масла, а на оси ординат—оптическую плотность, соответствующую каждому содержанию сивушного масла в анализируемом растворе.
Определение проводят не менее трех раз и из полученных значений находят среднее арифметическое.
Полученный градуировочный график используют для вычисления массовой концентрации сивушного масла в анализируемом растворе по величине оптической плотности.
7.5. Определение массовой концентрации сложных эфиров
Массовую концентрацию сложных эфиров в водке определяют фотоэлектроколориметрическим методом, который основан на измерении интенсивности окраски, полученной в процессе реакции хлорида железа с гидроксамовой кислотой, образующейся в результате взаимодействия сложных эфиров испытуемой водки с гидрохлоридом гидроксиламина:
|
|
Гидроксамовая кислота с железом образует комплексное соединение.
Реактивы: раствор гидрохлорида гидроксиламина концентрации 0,05моль/дм3: навеску гидрохлорида гидроксиламина массой 69,6 г растворяют в дистиллированной воде в мерной колбе вместимостью 500 см3, объем доводят до метки и перемешивают; раствор NaOH концентрацией 3,5моль/дм3: навеску NaOH массой 70,01 г растворяют в дистиллированной воде в мерной колбе вместимостью 500 см3, объем доводят до метки и перемешивают; раствор хлорида железа (III) концентрации 0,1 моль/дм3; раствор соляной кислоты концентрации 4 моль/дм3; раствор этилацетата в этиловом ректификованном спирте: к навеске этилацетата массой 25,0 г, взвешенной в склянке с пришлифованной пробкой, приливают небольшое количество этилового ректификованного спирта концентрацией 96,4 % об. сортов «Люкс» или «Экстра». Полученный раствор количественно переносят в мерную колбу на 500 см3, куда предварительно наливают 50 см3 этилового спирта сортов «Люкс» или «Экстра». Объем склянки доводят спиртом до метки и перемешивают.
Оборудование: фотоэлектроколориметр КФК-3.
Подготовка к анализу. Перед определением массовой концентрации сложных эфиров в водках, содержащих сахара, имеющие свободные альдегидную или кетонную группу (глюкоза, фруктоза, лактоза и др.), предварительно следует проводить перегонку, которую осуществляют по методике, изложенной в п.7.1.
Приготовление раствора реакционной смеси. Перед проведением анализа готовят раствор реакционной смеси путем смешивания равных объемов раствора гидрохлорида гидроксиламина и раствора гидроксида натрия, учитывая, что на проведение анализа одного образца испытуемого спирта расходуется 12 см3 смеси. Полученную смесь перемешивают и используют для анализа не позднее чем через 6 ч с момента приготовления.
Проведение анализа. Для проведения анализа требуется приготовить испытуемые растворы А и Б.
В две конические колбы вместимостью 50 см3 вносят по 6 см3 реакционной смеси. Затем в первую колбу приливают 3 см3 раствора соляной кислоты и перемешивают в течение 1 мин. Содержимое этой колбы именуют раствором Б, а содержимое второй колбы – раствором А.
В обе колбы приливают по 18 см3 анализируемой водки и одновременно осторожно перемешивают круговыми движениями в течение 2 мин.
Во вторую колбу с раствором А приливают 3 см3 раствора соляной кислоты и также перемешивают в течение 1 мин.
В обе колбы добавляют по 3 см3 раствора хлорида железа и одновременно перемешивают их содержимое вышеописанным образом в течение 1 мин.
Интенсивность образовавшейся окраски анализируемого раствора А измеряют в сравнении с раствором Б в кюветах с шириной рабочей грани 50 мм при светофильтре с длиной световой волны 540 нм. Полученную величину оптической плотности используют для расчета содержания сложных эфиров (Cэф, в мг/дм3 безводного спирта) по градуировочному графику, построенному по стандартным растворам этилацетата или по расчетному уравнению, выведенному на основании градуировочного графика.
Расчетное уравнение имеет вид: Cэф = D•100/ 0,0256•C ,
где D – оптическая плотность;
0,0303– постоянный коэффициент, полученный экспериментально;
С– объемная доля этилового спирта в испытуемом образце водки, %.
Построение градуировочного графика. В пять мерных колб вместимостью 200 см3 вносят соответственно 1; 2; 4; 6 и 8 см3 основного стандартного раствора этилацетата, объем доводят до метки этиловым спиртом сорта «Люкс» или «Экстра» и перемешивают. Получают рабочие стандартные растворы.
Полученные растворы используют для проведения реакции по вышеописанной методике. Оптическую плотность растворов измеряют в тех же условиях.
Одновременно проводят реакцию по приведенной методике с образцом этилового ректификованного спирта (безэфирного), использованного для приготовления стандартных растворов этилацетата.
Из значений оптических плотностей, измеренных после колориметрирования рабочих стандартных растворов, вычитают оптическую плотность, полученную после колориметрирования безэфирного образца этилового спирта. В результате получают значения оптических плотностей, соответствующие содержанию сложных эфиров 2,5; 5; 10; 15 и 20 мг/дм3.
На основании полученных данных строят градуировочную кривую, откладывая по оси абсцисс количество сложных эфиров в миллиграммах, а по оси ординат — соответствующую величину оптической плотности.
7.6. Определение объемной доли метилового спирта
Объемную долю метилового спирта в водке определяют визуально с помощью типовых спиртовых растворов или фотоэлектроколориметрическим методом, основанным на измерении интенсивности окраски в результате взаимодействия динатриевой соли хромотроповой кислоты (1,8–диокси-нафталин–3,6–дисульфокислота) с формальдегидом, образующимся в результате окисления метилового спирта, содержащегося в испытуемой водке, перманганатом калия.
Реактивы: раствор перманганата калия с массовой долей 1 %: навеску перманганата калия массой 5,0 г растворяют в 150 см3 дистиллированной воды в мерной колбе на 500 см3. Объем доводят до метки и перемешивают. Полученный раствор выдерживают в темноте в течение 2 суток; раствор динатриевой соли хромотроповой кислоты с массовой долей 10 %: навеску динатриевой соли хромотроповой кислоты массой 25 г растворяют в 15 см3 дистиллированной воды в мерной колбе вместимостью 25 см3, объем доводят водой до метки и перемешивают; 20 %- ный раствор сульфита натрия; концентрированная серная кислота.
Оборудование: фотоэлектроколориметр КФК-3.
Проведение анализа. В пробирку с пришлифованной пробкой вносят по 2 см3 раствора перманганата калия и испытуемой водки. Содержимое пробирки перемешивают и выдерживают при комнатной температуре в течение 3 мин. Затем вносят 0,4 см3 20%-ного раствора сульфита натрия для обесцвечивания реакционной смеси и 4 см3 концентрированной серной кислоты. Смесь тотчас же перемешивают и пробирку помещают в водяную баню с холодной водой. После охлаждения смеси до комнатной температуры (около 2 мин) в пробирку вносят по 0,1 см3 раствора динатриевой соли хромотроповой кислоты, содержимое перемешивают и пробирку помещают в кипящую водяную баню на 5 мин.
Далее пробирку охлаждают в бане до комнатной температуры и измеряют оптическую плотность раствора при светофильтрах с длиной световой волны 540 нм в кюветах с толщиной поглощающего свет слоя 10 мм. По полученной оптической плотности с использованием градуировочного графика определяют объемную долю метанола.
Построение градуировочного графика. Для построения графика используют стандартные типовые растворы с известным содержанием метилового спирта: 0,03; 0,05 и 0,13 % в 1 дм3 безводного спирта. Фотоэлектрометрический анализ этих растворов проводят аналогично выше описанному.
По полученным результатам строят градуировочный график, откладывая по оси абсцисс объемную долю метилового спирта (%), а по оси ординат — оптическую плотность.
Результаты исследований по п.п. 7.1-7.6 необходимо свести в таблицу и используя таблицу Приложения 4 сделать аргументированное заключение о качестве исследуемого образца водки.
Контрольные вопросы:
1. По каким показателям оценивается качество водок?
2. Дайте характеристику методам определения основных физико–химических показателей качества водок.
Раздел 4. КОНТРОЛЬ КАЧЕСТВА ВИНА И ВИНОМАТЕРИАЛОВ
Технохимический контроль качества вина включает как органолептическую оценку его качества, так и определение нормируемых физико–химических показателей, к которым относят: концентрацию спирта, сахаров, титруемых кислот, летучих кислот, диоксида серы, тяжелых металлов, приведенный экстракт и т.д.
Технохимическому контролю подвергается вся винодельческая продукция на всех стадиях технологического процесса. Контроль состоит в определении компонентов, входящих в сусло и вино, и заключении об их влиянии на качество вина.
Лабораторная работа № 8
Определение массовой концентрации сахаров
в вине и виноматериалах
Определение сахара в вине относится к числу основных, так как содержание сахара характеризует тип вина и его вкусовые особенности. Обычно в вине и винограде определяют содержание инвертного сахара. Иногда необходимо определять глюкозу и фруктозу в отдельности, а также сахарозу.
Глюкозу, фруктозу и их смесь относят к группе восстанавливающих (редуцирующих) сахаров, обладающих восстанавливающим действием в медно-щелочном растворе.
Массовую концентрацию редуцирующих сахаров определяют прямым методом. Метод основан на восстановлении инвертным сахаром, содержащимся в анализируемом изделии, оксида меди (II) до оксида меди (I). Определенный объем раствора Фелинга установленной концентрации титруют раствором анализируемого изделия, в котором предварительно проведена инверсия сахара, до полного восстановления оксида меди (II) в оксид меди (I). Диапазон измерения концентраций сахара 0–60,0 г/100 см3.
Реакции, лежащие в основе этого определения, можно выразить схемой:
|
|
|
|
|
|
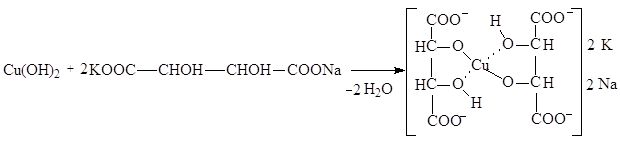
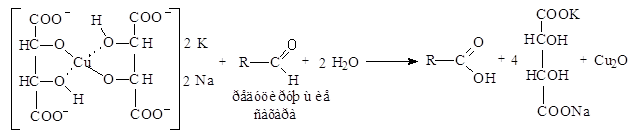
Оборудование: весы аналитические, секундомер, электроплитка, баня водяная, эксикатор, колбы мерные на 50, 100, 200, 250, 1000 см3, пипетки на 5, 10, 20, 25 см3, колбы конические, бюретки 25-50 см3 с делениями 0,1 см3, цилиндры.
Реактивы: медь (II) сернокислая 5-водная, калий-натрий виннокислый 4-водный, сахароза ч. д. а., кислота соляная, гидроокись натрия, фенолфталеин, метиленовая синь (индикатор), кальций хлористый обезвоженный чистый, сахар-рафинад, вода дистиллированная.
Приготовление раствора Фелинга I: взвешивают 69,39 г перекристаллизованной сернокислой меди, помещают в колбу вместимостью 1000 см3 , добавляют 500-700 см3 дистиллированной воды, перемешивают и фильтруют.
Приготовление раствора Фелинга (II): взвешивают 346 г виннокислого калия-натрия и переносят в колбу вместимостью 1000 см3 . Затем растворяют при слабом нагревании в 400-500 см3 дистиллированной воды.
Приготовление раствора гидроокиси натрия: взвешивают 103,2 г гидроокиси натрия, растворяют в 200 см3 дистиллированной воды. Полученный раствор переливают в колбу с виннокислым калием-натрием. Раствор в колбе вместимостью 1000 см3 доводят до метки дистиллированной водой и фильтруют.
Приготовление раствора метиленовой сини (индикатор) с массовой долей метиленовой сини 1 %: взвешивают 1,0 г метиленовой сини, переносят в мерную колбу вместимостью 100 см3 и растворяют в 50 см3 дистиллированной воды. Раствор доводят до метки дистиллированной водой.
Приготовление раствора гидроокиси натрия с массовой долей 20 %: 200 г гидроокиси натрия растворяют дистиллированной водой в мерной колбе на 1000 см3.
Приготовление раствора фенолфталеина с массовой долей 1% в растворе с концентрацией спирта 70%: 1,0 г фенолфталеина растворяют в 100 см3 ректификованного спирта с концентрацией 70 % по объему.
Подготовка к испытанию. Титр смеси растворов Фелинга I и II устанавливают следующим образом: сахарозу ч.д.а. или сахар рафинад, измельченный в пудру, выдерживают 2-3 дня в эксикаторе над хлористым кальцием. Навеску сахарозы или сахарной пудры 2-2,5 г тщательно смывают через воронку в мерную колбу вместимостью 250 см3 дистиллированной водой в объеме 50 см3. После растворения сахарозы в колбу добавляют 3 см3 соляной кислоты (плотность 1,19 г/см3), проводят инверсию сахарозы в течении 5 мин при температуре 67-70 °С. С инвертированным раствором проводят реакцию с Фелинговой жидкостью. Реакцию проводят три раза и берут среднее значение результатов, по которому вычисляют титр (Т) Фелинговой жидкости по формуле:
Т = V · m3/250
где V - объем раствора инвертного сахара, пошедший на титрование, см3;
m3 - масса навески сахарозы или сахарной пудры в г;
250 - объем колбы в см3.
Ход анализа
Проведение инверсии. Испытуемое изделие разбавляют дистиллированной водой в соответствии с таблицей.
Разведение вина в зависимости от содержания в нем сахара
|
Массовая концентрация сахара, г/100 см3 |
Объем испытуемого изделия, см3 |
Вместимость колбы, см3 |
| До 5 | — | — |
| 16-12 | 20 | 50 |
| 13-24 | 20 | 100 |
| 25-30 | 25 | 200 |
| 31-50 | 10 | 100 |
| 51-60 | 20 | 250 |
25 см3 разбавленного вина отмеривают пипеткой в мерную колбу вместимостью 100 см3. К раствору прибавляют 25 см3 дистиллированной воды, приливают 3 см3 соляной кислоты и содержимое колбы при частом перемешивании нагревают на водяной бане в течение 5 мин при температуре 67-70 °С. После этого раствор быстро охлаждают и нейтрализуют раствором гидроокиси натрия с массовой долей 20 % в присутствии фенолфталеина. После проведения инверсии и нейтрализации содержимое колбы при температуре 20 °С доводят дистиллированной водой до метки и перемешивают.
Проведение испытаний. В коническую колбу 150-200 см3 отмеривают по 10 см3 растворов Фелинга I и II и нагревают до кипения (отмерять пипетками с резиновыми грушами!). Затем из бюретки с пришлифованным краном осторожно и постепенно приливают инвертированный раствор до тех пор, пока синий цвет кипящей смеси не исчезнет почти полностью. После этого к смеси добавляют 2-5 капель раствора метиленовой сини с массовой долей 1 % и, не прекращая кипения, продолжают приливать испытуемый раствор по каплям пока цвет кипящей смеси не прейдет в красный или оранжевый.
Продолжительность кипения не должна превышать в течение всего титрования 3 минут, после чего отмечают количество израсходованного на титрование сахарного раствора, которое считают ориентировочным. Затем проводят повторное титрование, но к смеси растворов Фелинга I и II в колбу нагревания добавляют на 0,5-1,0 см3 испытуемого раствора меньше, чем пошло на первое титрование.
Смесь в колбе кипятят 2 мин и, не прекращая кипячение, добавляют 3-5 капель раствора метиленовой сини. Затем приливают из бюретки по 2-3 капли испытуемого раствора, давая смеси после каждого прибавления кипеть 2-3 сек до тех пор, пока синяя окраска в колбе не исчезнет и смесь не примет красную или оранжевую окраску. После этого отмечают количество израсходованного на титрование раствора.
Массовую концентрацию сахара (С3) в пересчете на сахарозу в г/100 см3 испытуемого раствора вычисляют по формуле:
С3 = Т ·100·n/ V1
где T - титр раствора Фелинга I и II;
п - коэффициент разведения;
V1- общий объем испытуемого раствора, пошедший на титрование и добавленный в реакционную смесь, см3 .
Массовую концентрацию сахара (С'з) в пересчете на инвертный сахар в г/100 см3 испытуемого раствора вычисляют по формуле:
С'з = Т ·100 · n ·1,05/ V1
где 1,05 - коэффициент перевода сахарозы в инвертный сахар.
За окончательный результат испытания принимают среднее арифметическое результатов двух параллельных определений, допускаемые расхождения между которыми не должны превышать 2.5%.
Контрольные вопросы:
1. Какие углеводы относят к редуцирующим сахарам?
2. Дайте характеристику состава и свойств редуцирующих сахаров.
3. Методы качественного и количественного определения представителей редуцирующих сахаров.
4. На чем основан прямой метод определения массовой концентрации сахаров?
5. Будет ли положительной реакция с растворами Фелинга для крахмала, сахарозы и мальтозы?
Лабораторная работа № 9
ОПРЕДЕЛЕНИЕ ОБЪЕМНОЙ ДОЛИ ЭТИЛОВОГО СПИРТА В ВИНЕ
(химический метод)
Этиловый спирт является основным продуктом виноделия. Это характерный для вина компонент, влияющий на его аромат и вкус. Этиловый спирт образуется в результате спиртового брожения виноградного сусла из сахаров. Из 1 г сахаров образуется 0,59–0,64% об. спирта. Для расчетов принят выход спирта 0,6%. Выход спирта зависит от исходного содержания сахаров в сусле, длительности брожения, расы дрожжей. В столовых винах этиловый спирт является фактором микробиальной стабильности.
Содержание этилового спирта в вине несколько снижается при его выдержке вследствие реакций окисления и этерификации, а также при технологических обработках. Для обеспечения требуемой крепости и формирования типа проводят спиртование этиловым спиртом крепленых и столовых (типа хереса) вин.
Объемная доля – это количество этилового спирта (см3), содержащегося в 100 см3 вина. Эта величина измеряется при температуре 20 °С и обозначается в процентах. Объемная доля спирта в винах различных типов варьирует от 9 до 20 % об.
Принцип метода. Метод основан на окислении спирта дихроматом калия в присутствии азотной кислоты до уксусной кислоты. Избыток дихромата калия оттитровывают после добавления йодида калия раствором тиосульфата натрия
Оборудование: установка для атмосферной перегонки; мерная коническая колба на 300 см3 с широким горлом (приемник); круглодонная колба на 100 см3; мерный цилиндр объемом 25 см3; бюретка объемом 50 см3 с ценой деления 0,1 см3.
Реактивы. Раствор хромата калия: 67,445 г К2СrO4 растворяют в воде и доводят объем до 1 дм3; азотная кислота концентрированная; раствор йодида калия: 300 г KI растворяют в воде, добавляют 100 см3 0,1 М раствора NaOH, доводят объем до 1 дм3; раствор тиосульфата натрия: 86,194 г Na2S2O3 5H2O растворяют в воде, добавляют 100 см3 0,1 М раствора NaOH и доводят до 1 дм3; раствор крахмала: 10 г растворенного в небольшом количестве воды крахмала вводят порциями в 500 см3 воды и кипятят, пока раствор не станет прозрачным, после охлаждения добавляют 500 см3 воды, в которой предварительно растворили 20 г KI и 10 см3 0,I M раствора NaOH.
Ход анализа. Собирают установку для атмосферной перегонки. В коническую колбу-приемник помещают 10 см3 раствора хромата калия и 25 см3 концентрированной HNO3 (в кислой среде хромат калия количественно переходит в дихромат). В перегонную колбу наливают 12 см3 воды, 1 см3 исследуемого вина и бросают 1-2 кусочка пемзы или пористого стекла для равномерного кипения. При содержании спирта в пробе больше 14 % об. вместо 1 см3 вина берут 0,5 см3. Для определения небольших количеств спирта (меньше 1,2 % об.) вместо 1 см3 берут 10 см3 исследуемой жидкости и добавляют вместо 12 только 3 см3 воды.
При введении исследуемой жидкости в перегонную колбу пипетка должна быть погружена в воду. Конец аллонжа погружают в приемную жидкость. Жидкость в перегонной колбе должна через 30 сек. закипеть, а еще через 30 сек. должны появиться капли дистиллята. Через 3-4 мин вынимают аллонж из дистиллята, промывают его водой и дистиллят разбавляют водой (приблизительно 300 см3). К дистилляту добавляют 10 см3 раствора KI и титруют раствором тиосульфата натрия до изменения окраски от коричневой до желтоватой. Добавляют 10 см3 крахмала и титруют до появления светло-голубой окраски.
Расчет. Объемную долю спирта (С, % об.) определяют по уравнению
|
С= 15,21 – 0,507 V, |
где 15,21 –максимально определяемое количество спирта, соответствующее 0 см3 раствора Na2S2O3, % об.;
0,507 – изменение концентрации спирта, соответствующее 1 см3 раствора Na2S2O3 % об.;
V – количество раствора Na2S2O3 ·5H2O, пошедшее на титрование, см3.
Если для анализа вместо 1 см3 образца брали 0,5 см3, то результаты определения умножают на 2. Соответственно, если объем пробы увеличивали в 10 раз или меньше, результат делят на кратность увеличения. Рекомендуется работать в области концентраций спирта от 0 до 14% об., когда на титрование расходуется от 30 до 2,4 см3 Na2S2O3.
Контрольные вопросы:
1. Методы определения объемной доли этанола в спиртных напитках;
2. Спиртовое брожение виноградного сусла. Факторы, влияющие на выход спирта;
3. На чем основано определение объемной доли спирта в вине химическим методом?
4. Причины снижения содержания спирта при выдержке вина.
Лабораторная работа № 10
Определение массовой концентрации летучих кислот в вине
Летучие кислоты являются показателем качества вина, обусловленным содержанием в нем алифатических одноосновных кислот с числом углеродных атомов от 1 до 9. Основным представителем летучих кислот вина является уксусная, составляющая 90 % от их общего содержания. Она образуется как вторичный продукт спиртового брожения сусла. Содержание летучих кислот лимитируется, так как они придают винам неприятный вкус и запах, и в высоких концентрациях свидетельствуют о микробиальных заболеваниях. Вина с повышенным содержанием летучих кислот могут быть исправлены путем сбраживания на мезге и обработки осадочными дрожжами.
Концентрация летучих кислот не должна превышать в белых винах 1,2 г/дм3, в красных - 1,5 г/дм3.
Принцип метода заключается в отгонке летучих кислот паром и определении их содержания в дистилляте титрованием гидроксидом натрия в присутствии фенолфталеина.
Оборудование. Установка для дистилляции паром, состоящая из парообразователя, перегонной колбы, холодильника и приемника. Можно использовать аппараты разных конструкций, удовлетворяющие следующим требованиям: из пара или воды, поступающих в перегонную колбу, должна быть удалена углекислота в такой степени, чтобы при добавлении к 250 см3 конденсата 0,1 см3 0,1 М раствора NaOH в присутствии 2 капель 1% раствора фенолфталеина появлялась розовая окраска, не исчезающая в течение 10 с; при перегонке водного раствора уксусной кислоты в дистиллят должно переходить ее не менее 99,5%. При перегонке 1 М раствора молочной кислоты в дистилляте не должно обнаруживаться более 0,5% этой кислоты.
Реактивы. Гидроксид натрия или калия 0,1 М раствор; фенолфталеин, 1% в 60-80% этиловом спирте; йод 0,005 М (0,01 н) раствор; крахмал, 1% раствор; тетраборат натрия (бура), насыщенный раствор; кислота винная; кислота соляная; баритовая или известковая вода.
Ход анализа. Из анализируемого вина удаляют углекислоту путем перемешивания в течение 2-3 мин в колбе, подключенной к водоструйному насосу. Парообразователь заполняют на 3/4 объема прозрачной баритовой или известковой водой. В перегонную колбу отмеряют пипеткой 10 см3 вина, добавляют около 0,25 г винной кислоты, закрывают колбу переходником, в который вмонтирована отводная трубка, соединяющая перегонную колбу с холодильником, включают нагревательный прибор и ведут перегонку до тех пор, пока в приемной колбе не соберется 100 см3 отгона. По окончании перегонки к дистилляту добавляют несколько капель фенолфталеина и титруют 0,1 М раствором щелочи.
Расчет. 1 см3 0,1 М раствора щелочи нейтрализует 0,006 г уксусной кислоты. Концентрация летучих кислот (С, г/дм3) определяется по формуле:
|
|
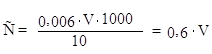
где V – количество щелочи, пошедшее на титрование, см3.
Для вин с содержанием
сернистой кислоты выше 50 мг/дм3 в результат определения вносят поправку на перешедшую
в дистиллят сернистую кислоту, свободную и связанную. Для этого по окончании
ацидиметрического титрования про![]() изводят йодометрическое определение
содержания SO2 в дистилляте. Оттитрованный раствор подкисляют каплей концентрированной соляной кислоты, прибавляют
5 см3 1% раствора крахмала и около 0,3 г йодида калия (на кончике
шпателя) и титруют 0,005 М раствором йода до появления синей окраски (свободная сернистая кислота).
изводят йодометрическое определение
содержания SO2 в дистилляте. Оттитрованный раствор подкисляют каплей концентрированной соляной кислоты, прибавляют
5 см3 1% раствора крахмала и около 0,3 г йодида калия (на кончике
шпателя) и титруют 0,005 М раствором йода до появления синей окраски (свободная сернистая кислота).
Для разрушения альдегидсернистого соединения в оттитрованный раствор прибавляют 20 см3 насыщенного раствора буры Na2B4O7. Если в течение 5 мин синяя окраска исчезает, то вносят 2-3 капли НС1 и вновь титруют 0,005 М раствором йода до ее повторного появления (связанная сернистая кислота).
Полный расчет содержания летучих кислот (С, г/дм3) в винах с учетом сернистой кислоты (в пересчете на уксусную кислоту) проводят по формуле:
|
|
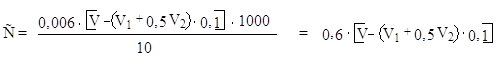
где 0,006 – количество уксусной кислоты, соответствующее 1 см3 0,1 М раствора NaOH, г;
V – количество 0,1 М раствора NaOH, израсходованное на титрование дистиллята, см3;
Vl – количество 0,005 М раствора йода, израсходованное на титрование свободной сернистой кислоты, см3;
V2 – количество 0,005 М раствора йода, израсходованное на титрование связанной сернистой кислоты, см3;
0,1 – коэффициент перевода 0,005 М раствора йода в 0,05 М раствор;
1000 – коэффициент пересчета на 1 дм3;
10 – количество вина, взятое для анализа, см3.
Контрольные вопросы:
1. Состав летучих кислот вина. Перечислите источники их образования;
2. Нормируемое содержание летучих кислот в белых и красных винах;
3. На чем основан метод определения содержания летучих кислот в вине?
4. В каких случаях и почему необходимо делать поправку на содержание сернистой кислоты?
Лабораторная работа № 11
Весовой метод определения содержания винной кислоты
в вине и виноматериалах
Винная кислота является двухосновной органической кислотой с двумя асимметрическими атомами углерода в молекуле, ее формула НООС–СН(ОН)–СН(ОН)–СООН. Винная кислота – кристаллы, хорошо растворимые в воде и спирте, плохо растворимые в эфире.
Известны 4 изомера винной кислоты; D–винная (виннокаменная) кислота, L–винная, D– и L–винные кислоты (виноградная) и оптически недеятельная мезовинная кислота.
Наибольшее значение имеет D–винная кислота, в основном содержащаяся в виноградной лозе. Это единственный источник получения винной кислоты в промышленном масштабе. В виноградных ягодах D–винная кислота накапливается в результате неполного окисления сахаров и составляет 0,2–1,0%.
Винная кислота и ее соли (кислый виннокислый калий, виннокислый кальций, виннокислый калий и др.) являются главными компонентами сусла и вина. Содержание винной кислоты в сусле 2,0–7 г/дм3, в вине – 2–5 г/дм3. В процессе приготовления и хранения вина ее содержание уменьшается вследствие выпадения в осадок в виде кислого виннокислого калия.
В сочетании с сахаром винная кислота и ее кислая калийная соль создают определенную вкусовую гармонию вина. Винная кислота и ее соли препятствуют развитию микроорганизмов. В то же время винная кислота и ее соли являются питательным субстратом для развития различных микроорганизмов, которые в процессе жизнедеятельности ее разлагают. Винная кислота образует комплексную железовинную соль, являющуюся катализатором окислительных процессов, необходимых при созревании вин. Содержание винной кислоты определялось до последнего времени колориметрическим методом, основанным на реакции винной кислоты с метаванадиевокислым аммонием.
В современной практике винная кислота определяется весовым методом при осаждении ее в виде рацемата кальция. Это определение может быть дополнено сравнительным объемным определением. Условия осаждения: рН, общий объем взятой пробы, концентрация ионов осадителя таковы, что осаждение рацемата кальция будет полным, тогда как D(–) тартрат кальция остается в растворе.
Если в вино была добавлена метавинная кислота, которая делает осаждение рацемата кальция неполным, необходимо подвергать ее предварительному гидролизу.
Метавинная кислота – смесь продуктов превращения D–винной кислоты при нагревании ее до температуры 170оС при пониженном давлении, среди которых преобладают моно– и диэфиры D–винной кислоты. Представляет собой твердый стекловидный продукт от бледно–желтого до темно–желтого цвета. В измельченном состоянии это белый или желтый кристаллический порошок. Очень гигроскопична, хорошо растворима в воде и этаноле. Используется для стабилизации вина от кристаллических помутнений, вызываемых калиевыми и кальциевыми солями винной кислоты.
Реактивы:
- раствор ацетата кальция, содержащий 10 г/дм3 Са: углекислый кальций СаСО3 (25 г), уксусная кислота (40 см3) (р20 = 1,05 г/ см3) и вода (до 1 дм3);
- рацемат кальция (СаС4Н4О6 . 4 Н2О): в цилиндрический стакан на 400 см3 вводят 20 см3 раствора L(+) винной кислоты (5 г/л), 20 см3 раствора D (–) виннокислого аммония (6,126 г/ дм3) и 6 см3 раствора ацетата кальция, содержащего 10 г/ дм3 Са. Оставляют для осаждения на 2 часа. Собирают осадок на стеклянном фильтре N 4, промывают в три приема приблизительно 30 см3 дистиллированной воды. Высушивают в шкафу при 70 °С до постоянного веса. Получают приблизительно 240 мг кристаллического рацемата кальция. Хранят в закрытой стеклянной емкости.
- раствор для осаждения (рН=4,75) : растворяют 150 мг D(–)виннокислого аммония в 900 см3 воды, добавляют 8,8 см3 раствора ацетата кальция, 5 мг рацемата кальция и доводят объем до 1 дм3. Взбалтывают в течение 12 ч. и затем отфильтровывают.
Примечание. Жидкость для осаждения может быть приготовлена также с помощью D(–) винной кислоты: кислота D(–) винная (122 мг), раствор гидроокиси аммония (р20 = 0,97 г/ см3), (25 г/100 см3) - 0,3 см3.
растворяют D(–) винную кислоту, добавляют гидроокись аммония, доводят объем приблизительно до 900 см3. Вводят 8,8 см3 раствора ацетата кальция, перемешивают, доводя рН до 4,75 добавлением уксусной кислоты, 5 мг рацемата кальция и доводят объем до 1 дм3. Взбалтывают в течение 12 ч и фильтруют.
Оборудование: стеклянный стакан на 600 см3, сушильный шкаф, стеклянный фильтр N 4, коническая колба на 50 см3.
Ход анализа.
В винах без метавинной кислоты. В цилиндрический стакан вместимостью 600 см3 вносят 500 см3 раствора для осаждения и 10 см3 вина. Перемешивают, очищают стенки стакана концом стеклянной палочки. Оставляют на 12 ч для осаждения (на ночь). Фильтруют через предварительно взвешенный стеклянный фильтр № 4, собирая осадок. Ополаскивают фильтратом стакан, где проводили осаждение, и извлекают последние частички осадка.
Высушивают в шкафу при 70 °С до постоянного веса, взвешивают; обозначают вес рацемата кальция СаС4Н4О6 , кристаллизованного с четырьмя молекулами воды, – р.
В винах с метавинной кислотой. Для вина с добавленной метавинной кислотой или при подозрении на ее добавление проводят гидролиз этой кислоты. В коническую колбу вместимостью 50 см3 помещают 10 см3 вина и 0,4 см3 чистой уксусной кислоты. Закрывают колбу пробкой с отводной трубкой и кипятят в течение 30 мин. После охлаждения переливают жидкость из конической колбы в цилиндрический стакан, ополаскивают колбу два раза водой по 5 см3 и продолжают определение по прописи, описанной выше. Расчет содержания метавинной кислоты производят так же, как и винной.
Одна молекула рацемата кальция соответствует половине молекулы L(+)– винной кислоты вина.
Количество винной кислоты на литр вина, выраженное в миллиграмм-эквивалентах, в граммах на литр и в граммах на литр кислого виннокислого калия, составляет соответственно 384,5 р, 28,84 р и 36,15 р с точностью до одного десятичного знака.
Контрольные вопросы:
1. Дайте характеристику строения и свойств изомеров винной кислоты.
2. Роль винной кислоты и ее солей в виноделии.
3. На чем основан весовой метод определения содержания винной кислоты?
4. Почему для осаждения винной кислоты необходимо предварительно гидролизовать метавинную кислоту?
Лабораторная работа № 12
Определение содержания основных органических кислот вина
Концентрация и соотношение органических кислот является важной характеристикой, несущей значительную информацию о процессах, проходящих в вине. В нем содержатся шесть основных органических кислот, играющих важную роль в формировании кислого вкуса вина. Винная, яблочная и лимонная кислоты переходят из винограда и обладают чисто кислым вкусом. Янтарная, молочная и уксусная кислоты образуются в результате спиртового или яблочно-молочного брожения.
12.1. Выделение органических кислот из вина с помощью
анионообменных смол
Оборудование. Колонка стеклянная, снабженная краном.
Реактивы: анионообменная смола вофатит 100-200 меш или АВ-17-8; 4-5% раствор (NH4)2CO3 или NH4HCO3; нитрат серебра, 0,1 М раствор; уксусная кислота, раствор 1 : 4: на 1 часть ледяной уксусной кислоты добавляют 4 части воды; уксусная кислота, 0,5, 6 и 30 % растворы; сульфат натрия, 0,5 М раствор: 71 г безводного Na2SO4 или 161 г Na2SO4 ·10 Н2О растворяют в мерной колбе объемом 1 дм3 и доводят водой до метки.
Подготовка колонки с ионообменной смолой. Органические кислоты выделяют из вина с помощью анионообменной смолы вофатит 100-200 меш или АВ-17-8 в уксуснокислой форме. Препарат анионита в С1–-форме промывают 4-5% раствором (NH4)2CO3 или NH4HCO3, потом водой до исчезновения реакции на хлорид-ионы (добавление к 1 см3 промывной воды 1-2 капель 0,1М AgNO3 не должно вызывать помутнения). Анионит промывают 2-3 раза водой, потом теплым (40-50°С) раствором уксусной кислоты 1:4 до исчезновения вспенивания. Для перевода анионита в уксуснокислую форму к 100 г смолы приливают 200 см3 30 %-ной уксусной кислоты и оставляют в контакте не менее суток при периодическом перемешивании. Смолу хранят в растворе 6% уксусной кислоты.
Ионообменные стеклянные колонки (высотой 15 см, диаметром 0,8-1,0см) заполняют анионитом (высота слоя 5-6 см). Для предупреждения возможного вытекания суспензии смолы или ее взмучивания на дно колонки помещают ватный тампон и таким же тампоном прикрывают слой смолы сверху.
Следят за тем, чтобы смола в колонке все время находилась под слоем жидкости.
Ход работы. Заполненную колонку 3-4 раза промывают 0,5 % раствором уксусной кислоты порциями по 10 см3, пропускают через нее 10 см3 исследуемой пробы (сусло или вино) со скоростью 1 см3/мин и снова промывают 10 см3 0,5 % раствора уксусной кислоты и 7 раз по 10 см3 дистиллированной воды (скорость вытекания жидкости 2 см3/мин). Адсорбированные кислоты элюируют 0,5 М раствором Na2SO4. Элюат собирают в мерную колбу объемом 50 см3 до метки, тщательно перемешивают и используют для определения массовой концентрации кислот: винной (п.12.2 ), молочной (п.12.3) и яблочной (п.12.4).
12.2. Колориметрический метод определения массовой концентрации
винной кислоты
Оборудование: Спектрофотометр.
Реактивы: Метаванадат аммония 2%-ный раствор: 10 г NH4VO3 растворяют в 150 см3 1 М NaOH в мерной колбе объемом 500 см3, добавляют 200 см3 27% раствора ацетата натрия и доводят водой до метки; 1 М (2 н) и 0,05 М (0,1 н) растворы серной кислоты; 0,05 М раствор йодной кислоты (НIO4-2Н2О); 10% раствор глицерина; стандартный раствор винной кислоты (0,5 мг/см3): 250 мг винной кислоты растворяют в мерной колбе объемом 500 см3.
Ход анализа. По 10 см3 элюата, полученного в работе п. 12.1, помещают в 2 колбы объемом 50 см3. В колбу I добавляют 1 см3 1 М раствора H2SO4, 2,5 см3 0,05 М раствора H2SO4 и 0,5 см3 раствора глицерина. В колбу II (контрольный раствор) помещают 1 см3 1 М раствора H2SO4, 2,5 см3 йодной кислоты, дают постоять 15 мин до полного разрушения винной кислоты, добавляют 0,5 см3 10 %-ного раствора глицерина для удаления избытка периодата и оставляют на 2 мин. Добавляют сначала в колбу II, а потом в колбу I по 2,5 см3 раствора метаванадата аммония и измеряют ровно через 90 с оптическую плотность раствора в колбе I против контрольного раствора при длине волны 490 нм в кювете толщиной 5 мм.
Концентрацию винной кислоты определяют по калибровочному графику с учетом разбавления при обработке анионитом (разбавление в данных условиях определения равно 5).
Построение калибровочного графика. 10, 20, 30, 40 и 50 см3 стандартного раствора винной кислоты пропускают через ионообменные колонки, собирая по 50 см3 элюата. Растворы содержат винной кислоты соответственно 0,1, 0,2; 0,3; 0,4; 0,5 г/дм3. Отбирают 2 раза по 10 см3 каждого из элюатов и анализируют, как указано выше. Строят график зависимости оптической плотности от концентрации винной кислоты.
Контрольные вопросы:
1. Дайте характеристику строения и свойств изомеров винной кислоты.
2. Роль винной кислоты и ее солей в виноделии.
3. На чем основан калориметрический метод определения содержания винной кислоты?
12.3. Определение массовой концентрации молочной кислоты
Молочная кислота НООС–СНОН–СН3 относится к одноосновным алифатическим оксикислотам винограда и вина. Основное количество молочной кислоты образуется в процессе яблочно-молочного брожения, которое позволяет смягчить резкий вкус «зеленой» кислотности молодых вин. Малокислотные столовые вина с остаточным сахаром, а также крепкие и десертные вина иногда подвергаются молочнокислому брожению, которое сопровождается повышением содержания молочной и летучих кислот. Заболевание вина сопровождается появлением «квашенных» тонов и вкуса молочной сыворотки, иногда «мышиного» привкуса. Концентрация молочной кислоты в белых винах может достигать 2,5 г/дм3, в красных - 4,5 г/дм3.
Принцип метода. Метод основан на окислении молочной кислоты сульфатом церия (IV) в ацетальдегид, который, реагируя с пиперидином и нитропруссидом натрия, дает окрашенный продукт, определяемый колориметрически.
Оборудование. Фотоэлектроколориметр, пробирки с притертой пробкой объемом 25 см3; водяная баня с термостатом на 65°С.
Реактивы. Сульфат церия Ce(SO4)2 4H2O, 0,1 М раствор в серной
кислоте: ![]() 40,431 г Ce(SO4)2 · 4H2O растворяют в 350 см3
1 М (2 н) H2SO4 в мерной колбе объемом 1 дм3 и доводят до метки;
пиперидин: 200 см3 пиперидина разбавляют водой до 1 дм3
(готовят за 3-4 дня до применения); нитропруссид натрия, 0,4% раствор: 1 г
реактива растворяют в воде в мерной колбе объемом 250 см3 (готовят
перед применением); молочная кислота, 1 М раствор.
40,431 г Ce(SO4)2 · 4H2O растворяют в 350 см3
1 М (2 н) H2SO4 в мерной колбе объемом 1 дм3 и доводят до метки;
пиперидин: 200 см3 пиперидина разбавляют водой до 1 дм3
(готовят за 3-4 дня до применения); нитропруссид натрия, 0,4% раствор: 1 г
реактива растворяют в воде в мерной колбе объемом 250 см3 (готовят
перед применением); молочная кислота, 1 М раствор.
Ход анализа. К 5 см3 элюата, полученного в работе 12.1, наливают в пробирку объемом 25 см3, добавляют 5 см3 раствора сульфата церия, закрывают и оставляют на 90 мин при комнатной температуре. Затем добавляют 5 см3 раствора пиперидина, размешивают, фильтруют через складчатый фильтр. К 5 см3 фильтрата добавляют 5 см3 раствора нитропруссида натрия. Размешивают и переносят сразу в кювету толщиной 1 см для колориметрирования. Интенсивность окраски определяют при длине волны 570 нм в кювете толщиной 0,5 см против воды. Максимальная окраска достигается через 60-90 с. За это время делают 2-3 замера оптической плотности. Из значения максимальной плотности окраски вычитают значение оптической плотности контрольного опыта (вместо 5 см3 элюата берут 5 см3 раствора сульфата натрия, анализируют в тех же условиях, что и элюат).
Содержание молочной кислоты (в г/дм3) определяют по калибровочному графику с учетом разбавления пробы при обработке (разбавление равно 5).
Построение калибровочного графика. В мерную колбу объемом 100 см3 наливают 1 см3 1 М раствора молочной кислоты и доводят 7,1% раствором сульфата натрия до метки (рабочий раствор). В мерные колбы объемом 50 см3 помещают по 2,5; 5,0; 7,5; 10,0; 12,5 и 15 см3 рабочего раствора и доводят раствором сульфата натрия до метки. Концентрация молочной кислоты в растворах составляет 0,045; 0,090; 0,135; 0,180; 0,225; 0,270 г/дм3. Из каждой колбы отбирают по 5 см3 и анализируют, как описано ранее. Строят график зависимости оптических плотностей от концентраций.
Примечание. Вина, содержащие более 250 мг/дм3 диоксида серы, могут содержать некоторое количество альдегидсернистой кислоты, которая определяется так же, как молочная кислота. В этом случае в результат определения необходимо внести поправку. Для этого 15 см3 элюата смешивают в пробирке с притертой пробкой с 5 см3 ацетата натрия массовой концентрации 27 г/100 см3 и 2 см3 раствора H2SO4 (77,5 мл H2SO4 доводят до 100 см3 водой). Затем добавляют 5 см3 раствора нитропруссида натрия (2 г/100 см3) и 5 см3 10% раствора пиперидина. После смешивания измеряют оптическую плотность при соблюдении условий, описанных для измерения молочной кислоты. По этой величине на калибровочном графике находят концентрацию В (г/дм3) молочной кислоты совместно с альдегидсернистой кислотой. Если L' - содержание молочной кислоты в вине без корректировки, то реальное содержание молочной кислоты (L, г/дм3) составит
| L = L' – 0,4 В. |
Контрольные вопросы:
1. Источники образования молочной кислоты в винах;
2. Роль молочной кислоты в виноделии;
3. На чем основан метод определения содержания молочной кислоты?
12.4. Определение массовой концентрации яблочной кислоты
Яблочная кислота относится к многоосновным оксикислотам, содержит асимметрический углеродный атом. В процессе созревания винограда количество ее уменьшается и в период физиологической зрелости ягод составляет 2–5 г/кг. В процессе спиртового брожения концентрация яблочной кислоты снижается вследствие ее потребления дрожжами. Молочнокислые бактерии сбраживают яблочную кислоту в молочную, при этом происходит снижение содержания титруемых кислот и повышение рН, формирование мягкого гармоничного вкуса вин. Концентрация яблочной кислоты в винах не превышает 5 г/дм3.
Значительное количество яблочной кислоты содержат незрелые ягоды: до 15 г на 1кг винограда. Яблочная кислота участвует в дыхательных процессах и к моменту достижения технической зрелости ее содержание снижается до 2–5 г на 1кг. В северных районах виноградарства, а также при холодной погоде осенью в южных районах виноград может быть излишне кислым из-за избытка яблочной кислоты. Столовые вина из такого винограда имеют привкус так называемой "зеленой кислотности". Под действием дрожжей и бактерий при благоприятных условиях происходит биологическое кислотопонижение, связанное с превращением яблочной кислоты в слабо диссоциированную молочную кислоту. Применяют также химические методы нейтрализации избыточного количества яблочной кислоты в виноградном сусле или вине.
Принцип метода. Метод основан на реакции взаимодействия яблочной кислоты с хромотроповой и серной кислотами, вследствие чего образуется комплекс желто-зеленого цвета, интенсивность окраски которого определяется колориметрически.
Оборудование. Фотоэлектроколориметр, пробирки с притертой пробкой объемом 25 см3.
Реактивы. Серная кислота концентрации 96% мас. и 86% мас; динатриевая соль хромотроповой кислоты (хромотроповая кислота – это 1,8–ди–оксинафталин–3,6–дисульфокислота), 5% водный раствор (готовят непосредственно перед определением, хранят в темной склянке); стандартный раствор DL- или L- яблочной кислоты (1 мг/см3): 500 мг яблочной кислоты растворяют в мерной колбе объемом 500 см3; сульфат натрия, 7,1%-ный раствор.
Техника определения. 1 см3 элюата, полученного в работе 12.1, помещают в пробирку с притертой пробкой, добавляют 1 см3 раствора соли хромотроповой кислоты и 10 см3 96% H2SO4, размешивают и погружают в кипящую водяную баню на 20 мин. Быстро охлаждают до 20°С и точно через 90 мин определяют оптическую плотность раствора. Все операции с момента прибавления динатриевой соли хромотроповой кислоты следует проводить в затемненном месте. Колориметрируют при длине волны 420 нм в кювете толщиной 10 мм.
Раствор сравнения готовят так же, заменяя 1 см3 элюата на такое же количество 7,1%-ного раствора Na2SO4 и 96% серную кислоту - на 86%.
Концентрацию яблочной кислоты определяют по калибровочному графику. Полученный результат умножают на разбавление (в данных условиях разбавление равно 5).
Построение калибровочного графика. Через 6 ионообменных колонок, заполненных анионитом, пропускают 5, 10, 15, 20, 25 и 30 см3 стандартного раствора яблочной кислоты и выполняют все операции по промывке и элюированию. По 1 см3 элюатов отбирают в пробирки и анализируют, как указано ранее. Строят график зависимости оптической плотности от концентрации яблочной кислоты. Концентрация элюатов соответствует концентрации яблочной кислоты 0,1; 0,2; 0,3; 0,4; 0,5; 0,6 г/дм3 .
Контрольные вопросы:
1. Источники образования яблочной кислоты в винах;
2. Роль яблочной кислоты в виноделии;
3. На чем основан метод определения содержания яблочной кислоты?
12.5. Определение массовой концентрации лимонной кислоты
Лимонная кислота НООС–СН2–С(СООН)(ОН)–СН2–СООН относится к группе многоосновных оксикислот. Содержится в небольших количествах (0,2–0,5 г/кг) в ягодах винограда, а также образуется как вторичный продукт при спиртовом брожении. Содержание в винах составляет до 0,3 г/дм3. В виноделии разрешено использовать лимонную кислоту для исправления низкокислотных виноматериалов: увеличения содержания титруемых кислот и обеспечения требуемых кондиций, а также обеспечения стабильности к микробиальным помутнениям. Для этой цели лимонная кислота вводится в вино в количестве, не превышающем 2 г/дм3.
Введение лимонной кислоты в вина, в которых идет процесс яблочно-молочного брожения, нецелесообразно, так как бактерии ее ассимилируют.
Лимонная кислота применяется также для предотвращения железного касса, поскольку образует стойкий растворимый комплекс с ионами трехвалентного железа.
Принцип метода. Лимонную кислоту фиксируют вместе с другими органическими кислотами вина на анионообменной смоле. Затем проводят фракционное элюирование, которое позволяет отделить ее от лимонно-яблочной кислоты. Лимонная кислота с помощью щадящего окисления переводится в ацетон, который отделяют дистилляцией. Уксусный альдегид, отгоняемый вместе с ацетоном, окисляется до уксусной кислоты, после чего ацетон определяют иодометрическим методом.
Оборудование: анионообменная колонка, установка для атмосферной перегонки.
Реактивы: Смола Дауэкс 1×2 (50-100 меш.); уксусная кислота, растворы 4 М и 2,5 М; гидроксид натрия, раствор 2 М; серная кислота (р20=1,84 г/см3), разбавленная 1:5 и 1:3 по объему; буферный раствор рН 3,2-3,4: 150 г дигидро-фосфата калия КН2РО4, 5 см3 фосфорной кислоты (р20= 1,70 г/см3) доводят водой до 1 дм3; сульфат марганца, раствор массовой концентрации 50 г/дм3; пемза; перманганат калия, растворы 0,01 М и 0,4 М; сульфат железа (II) FeSO4∙7H2O, раствор массовой концентрации 40г/100 см3; гидроксид натрия, 5 М раствор; йод, 0,01 М раствор; тиосульфат натрия Na2S2O3, 0,02 М раствор; раствор крахмала.
Подготовка анионообменной колонки: в бюретку объемом 25 см3 с краном помещают тампон из стекловаты и наливают 20 см3 смолы Дауэкс 1×2. Вначале подвергают смолу двум полным циклам регенерации с попеременным пропусканием растворов 1 М соляной кислоты и гидроксида натрия. Ополаскивают 50 см3 воды (пропускание раствора гидроксида натрия вызывает уплотнение с последующим набуханием при промывании водой, что мешает стеканию). Как только первые миллилитры воды начинают проходить через колонку, рекомендуется взбалтывать смолу, чтобы поднять ее со дна бюретки. Переводят смолу в ацетатную форму, пропуская 250 см3 4 М раствора уксусной кислоты; промывают 100 см3 воды.
Анализируемый образец пропускают через колонку в соответствии с описанием, приведенным далее. После элюирования кислот промывают смолу 50 см3 дистиллированной воды и производят снова насыщение смолы 4 М раствором уксусной кислоты. Ополаскивают 100 см3 дистиллированной воды.
Ход анализа:
Отделение лимонной и лимонно-яблочной кислот. Пропускают 25 см3 вина через анионообменную колонку Дауэкс 1×2 в ацетатной форме со скоростью 1,5 см3 в мин. Ополаскивают колонку 20 см3 дистиллированной воды в три приема. Элюируют кислоты 200 см3 2,5 М раствора уксусной кислоты, пропуская элюат с той же скоростью. В этой фракции элюата содержатся кислоты: янтарная, молочная, галактуроновая, лимонно-яблочная и почти вся яблочная. Затем производят элюирование лимонной и винной кислот, пропуская через колонку 100 см3 2 М раствора гидроксида натрия, собирают элюат в колбу прибора.
Окисление. В двухгорловую круглодонную колбу на 500 см3, содержащую второй элюат, добавляют серную кислоту, разбавленную 1:5 для доведения рН до 3,2-3,8. Затем добавляют 25 см3 буферного раствора, 1 см3 раствора сульфата марганца и несколько зерен пемзы. Доводят до кипения и отгоняют 50 см3, которые отбрасывают. Помещают 0,01 М раствор перманганата калия в воронку с краном и вводят его со скоростью 1 капля в секунду в кипящий элюат. Дистиллят собирают в склянку объемом 500 см3 с притертой пробкой, содержащую несколько см3 воды. Продолжают окисление до окрашивания жидкости в коричневый цвет, указывающий на избыток перманганата.
Отделение ацетона. Если объем дистиллята меньше 90 см3, его доводят до этого объема дистиллированной водой, добавляют 4,5 см3 разбавленной 1:3 серной кислоты и 5 см3 0,4 М раствора перманганата калия. Если объем собранного дистиллята значительно превышает 90 см3, доводят его до 180 см3 и удваивают дозы добавляемых реактивов. В этих условиях (в среднем 0,25 М серной кислоты и 0,02 М перманганата калия) уксусный альдегид окисляется в уксусную кислоту, тогда как ацетон остается без изменения.
Сосуд закупоривают и выдерживают в течение 45 мин при комнатной температуре. По окончании этого времени избыток перманганата калия разрушают добавлением раствора сульфата железа (II). Дистиллируют и собирают приблизительно 50 см3 дистиллята в сосуд со шлифом, содержащий 5 см3 5 М раствора гидроксида натрия.
![]() Определение ацетона. Добавляют к содержимому
колбы 25 см3 0,01 М раствора йода, выдерживают 20 мин. Этого
количества достаточно при содержании лимонной кислоты не более 0,5–0,6 г/см3;
для более высоких концентраций указанный
объем раствора йода недостаточен, и раствор не приобретает желтую
окраску, характерную для избытка йода. В этом случае удваивают или утраивают
количество добавляемого йода до получения чистой желтой окраски. Однако в исключительных случаях, когда
содержание в вине лимонной кислоты превышает 1,5 г/ дм3,
предпочтительно начать анализ снова с 10 см3 вина. Затем добавляют 8
см3 разбавленной 1: 5 серной кислоты и титруют избыток йода 0,02 М
раствором тиосульфата натрия в присутствии крахмала. Количество (см3)
раствора тиосульфата натрия, израсходованного на титрование, обозначают V.
Определение ацетона. Добавляют к содержимому
колбы 25 см3 0,01 М раствора йода, выдерживают 20 мин. Этого
количества достаточно при содержании лимонной кислоты не более 0,5–0,6 г/см3;
для более высоких концентраций указанный
объем раствора йода недостаточен, и раствор не приобретает желтую
окраску, характерную для избытка йода. В этом случае удваивают или утраивают
количество добавляемого йода до получения чистой желтой окраски. Однако в исключительных случаях, когда
содержание в вине лимонной кислоты превышает 1,5 г/ дм3,
предпочтительно начать анализ снова с 10 см3 вина. Затем добавляют 8
см3 разбавленной 1: 5 серной кислоты и титруют избыток йода 0,02 М
раствором тиосульфата натрия в присутствии крахмала. Количество (см3)
раствора тиосульфата натрия, израсходованного на титрование, обозначают V.
В аналогичных условиях проводят контрольный опыт, заменив 50 см3 дистиллята 50 см3 дистиллированной воды. Обозначают количество раствора тиосульфата натрия (см3) V`.
Расчет. 1 см3 0,01 М раствора йода соответствует 0,64 мг лимонной кислоты. В данных условиях количество лимонной кислоты (С, мг/дм3) определяется выражением:
|
С = 25,6 (V` – V). |
Концентрацию лимонной кислоты округляют до целого значения.
Контрольные вопросы:
1. Источники образования лимонной кислоты в винах;
2. С какой целью и в каком количестве добавляют лимонную кислоту в вино?
3. На чем основан метод определения содержания лимонной кислоты?
Раздел 5. КОНТРОЛЬ КАЧЕСТВА БЕЗАЛКОГОЛЬНЫХ НАПИКОВ
Лабораторная работа № 13
Определения количества цитрусовых эфирных масел
в безалкогольных напитках
В качестве ароматических веществ, вносимых в безалкогольные напитки, используют эссенции, настои, а также спиртовые растворы эфирных масел и душистых веществ.
Эфирные масла – это масло лавра благородного, а также розовое, эвкалиптовое, лимонное и мандариновое масла. Для выделения эфирных масел используют перегонку с паром. Лимонное эфирное масло получают из цедры лимона, по внешнему виду это легкоподвижная жидкость желтого или желто–коричневого цвета с запахом лимона. Мандариновое эфирное масло – легкоподвижная жидкость желтого или желто–коричневого цвета, как и лимонное масло, только с запахом мандарина. Основным компонентом эфирных масел является моноциклический терпен – лимонен:
|
|
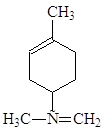
Предлагаемый метод определения эфирных масел в безалкогольных напитках основан на количественном соединении α-лимонена с бромом, при этом излишек брома обесцвечивается индикатором.
Приборы: весы лабораторные аналитические; весы лабораторные технические; бюретка на 25 см3 с ценой деления 0,1 см3; колбы мерные на 100, 500, 1000 см3; пипетки на 5, 20, 50 см3; колбы конические 250 и 750 см3; цилиндр мерный 500 см3; капельница стеклянная лабораторная; секундомер.
Реактивы: калий бромноватокислый; натрий серноватистокислый 0,1 н стандарт-титр; калий йодистый; соляная кислота концентрированная; крахмал растворимый; спирт этиловый ректификованный; метиловый оранжевый водный раствор с массовой концентрацией 1,0 г/дм3; вода дистиллированная.
Подготовка к анализу:
1. Приготовление раствора калия бромноватокислого с молярной концентрацией 0,02 моль/дм3: 0,560 г калия бромноватокислого растворяют в мерной
колбе водой и доводят объем до 1 дм3.
2. Приготовление раствора тиосульфата натрия с молярной концентрацией 0,02 моль/дм3: из фиксанала готовят раствор тиосульфата натрия с молярной концентрацией 0,1 моль/дм3, который разбавляют в 5 раз.
3. Приготовление раствора крахмала с массовой долей 1%: в химический стакан вместимостью 250 см3 наливают 80 см3 дистиллированной воды и доводят до кипения. 1 г растворимого крахмала растворяют в 20 см3 дистиллированной воды и приливают в кипящую воду при непрерывном помешивании. Раствор крахмала доводят до кипения и охлаждают.
4. Установление поправочного коэффициента на молярность раствора калия бромноватокислого с молярной концентрацией 0,02 моль/дм3 по раствору натрия серноватистокислого с молярной концентрацией 0,02 моль/дм3 : в конической колбе вместимостью 750 см3 растворяют 0,5 г йодистого калия в 100 см3 воды. К содержимому колбы пипеткой добавляют 5 см3 концентрированной соляной кислоты, 20 см3 раствора бромноватокислого калия с молярной концентрацией 0,02 моль/дм3 и 400 см3 воды. Полученную смесь перемешивают и через 2–3 мин оттитровывают выделившийся йод раствором тиосульфата натрия с молярной концентрацией 0,02 моль/дм3. В процессе титрования содержимое колбы постоянно взбалтывают. При появлении слабожелтого окрашивания раствора добавляют 1—2 см3 раствора крахмала массовой доли 1% и продолжают титровать до изменения окрашивания из синего в бесцветное. Проводят два параллельных определения.
Ход анализа.
Титрование напитка. В коническую колбу вместимостью 250 см3 пипеткой отбирают 50 см3 хорошо перемешанного в бутылке напитка, прибавляют пипеткой 50 см3 этилового спирта (95—96% об.), 5 см3 соляной кислоты, разведенной на 1 часть концентрированной кислоты 2 части дистиллированной воды. Смесь взбалтывают и добавляют 3 капли индикатора метилового оранжевого.
Титрование медленное при постоянном энергичном взбалтывании со скоростью 0,1 см3 раствора калия бромноватокислого в каждые 30 секунд до исчезновения розового окрашивания. Для сравнения окраски рядом ставят такую же колбу с тем же напитком, спиртом и соляной кислотой в заданных выше количествах.
Контрольное титрование. Общий расход калия бромноватокислого суммируется из расхода на титрование спирта и компонентов напитка. Титрование спирта проводят аналогично, но с заменой напитка дистиллированной водой.
Титрование проводится при использовании на анализ каждой новой партии спирта и свежеприготовленных реактивов.
Расход раствора калия бромноватокислого на титрование компонентов напитка в зависимости от наименования приведен в таблице.
| Наименование напитка |
Объем раствора калия бромноватокислого с молярной концентрацией 0,02 моль/дм3 на титрование компонентов, см3 |
| Апельсин | 0,55 |
| Колокольчик | 0,25 |
| Лимон | 0,45 |
| Мандариновый | 0,60 |
| Цитрусовый | 0,40 |
| Экстра Ситро | 0,70 |
| Апельсиновый для больных диабетом на ксилите | 0,15 |
| Лимонный для больных диабетом на ксилите | 0,15 |
| Лимонный негазированный | 0,45 |
| Саяны тонизирующий | 0,60 |
Массовую долю (X) эфирных масел (в %) вычисляют по формуле:
(n–(no + B)) . K . 0,000623 . 100
Х = –––––––––––––––––––––––––––––––– ;
a . 0,90 . 0,85
где: n – объем раствора калия бромноватокислого, расходуемого на титрование взятого объема напитка, см3;
no –объем раствора калия бромноватокислого, расходуемого на титрование спирта, см3;
В–объем раствора калия бромноватокислого, расходуемого на титрование компонентов, входящих в напиток, согласно таблице;
К – коэффициент нормальности раствора калия бромноватокислого;
а – объем пробы напитка, взятый на титрование, см3;
0,90 – коэффициент перевода титруемого компонента на массу эфирного масла;
0,85 – плотность эфирного масла, г/см3;
0,000623 – масса титруемого компонента эфирного масла, в г, которое соответствует 1 см3 раствора калия бромноватокислого с молярной концентрацией 0,02 моль/дм (для апельсинового масла 0,00062, мандаринового – 0,00061, лимонного – 0,00064).
При соблюдении условий изложенной выше прописи проведения анализа, формула расчета количества эфирных масел в напитках может быть сокращена до следующего вида: массовая доля X = n - (nо + В) . К . 0,00163.
За окончательный результат испытаний принимают среднее арифметическое результатов двух параллельных определений, допускаемое расхождение между которыми не должно превышать 0,1 см3 раствора калия бромноватокислого с молярной концентрацией 0,02 моль/дм3 .
Контрольные вопросы:
1. Какие ароматические вещества, используются в производстве безалкогольных напитков?
2. На чем основан метод определения содержания цитрусовых эфирных масел?
Лабораторная работа № 14
Получение колера для напитков
В производстве безалкогольных напитков с целью придания соответствующего цвета продукции используют красители, которые делятся на натуральные и естественные (синтетические). К натуральным красителям относятся колер, энокраситель, а также красители, получаемые из ягод бузины, выжимок черники, кизила, вишни и др. К синтетическим красителям относятся тетразин Ф и краситель индигокармин.
Колер представляет собой карамелизованный сахар, обычно — карамелизованную сахарозу. Энокраситель получают из выжимок винограда красных сортов. Основным красящим веществом энокрасителя является энин, входящий в группу антоцианинов.
Метод получения колера заключается в нагревании сахарозы при температуре, близкой к температуре плавления (185–190 °С), что вызывает ее глубокие химические изменения – обезвоживание, карамелизацию, гидролиз, таутомерные и изомерные превращения моноз, ангидридизацию и оксиметилфурфурольное разложение, полимеризацию. В результате пиролиза образуется сложная смесь, состоящая из ангидридов различных сахаров, производных фурана, кислот жирного ряда, темноокрашенных (гуминовых) соединений и других веществ.
Для получения колера кроме сахара используют кристаллическую глюкозу, крахмальную патоку, а также смесь углеводов и аминокислот. В последнем случае происходят меланоидиновые реакции. Известно много сортов колера, например только фирма May–Lambert выпускает их свыше 70, различающихся интенсивностью окраски, вязкостью, рН, изоэлектрической точкой и другими свойствами.
Колер добавляют во многие напитки, которые должны иметь цвет от светло-желтого до темно–коричневого. Обычная норма колера невелика - от 2 до 7 кг на 1000 дал купажа. Несколько больше его идет в «Шоколадный крем» (36 кг) и некоторые настойки («Хинная», «Охотничья», «Перцовка»). Особенно в больших количествах (1300 кг) его добавляют в Рижский чёрный бальзам. Бальзаму при этом он не только придает соответствующую окраску, но и участвует в формировании вкуса, аромата и физико-химических свойств напитка.
Практически во все сорта темного пива добавляют колер, что обеспечивает им темно-коричневый цвет и сладкий вкус. Его также используют при приготовлении безалкогольных напитков. Перед внесением в купаж колер разбавляют водой в соотношении 1:1.
По внешнему виду колер вязкая густая жидкость темно–коричневого цвета, горького вкуса.
Приборы и реактивы: электрический сушильный шкаф, весы лабораторные общего назначения с наибольшим пределом взвешивания 200 г, стаканы фарфоровые или из термостойкого стекла вместимостью 500 – 1000 см3, колбы, воронка, вода дистиллированная, ареометры с пределами измерения плотности 1,2 – 1,5, термометры лабораторные на интервал температур 50 – 200 °С, цилиндры.
Ход работы: Для предохранения от брызг горячего колера необходимо работать в фартуках и защитных очках! В фарфоровый или стеклянный стакан загружают 100 г сахара (с точностью до 0,5 г). Стакан подбирают с таким расчетом, чтобы сахар занимал 30 – 35 % полезной емкости стакана. Затем стакан ставят в сушильный шкаф, предварительно нагретый до 80 – 100 °С, нагревают при периодическом перемешивании 1 – 2 мин с интервалом 5 – 10 мин. При температуре 160 °С сахар расплавляется и постепенно буреет. Когда весь сахар расплавится, температуру постепенно повышают до 175 – 180 °С и поддерживают ее 15 – 20 мин при перемешивании массы. При готовности колера нагрев прекращают, а перемешивание продолжают 10 – 15 мин, после чего подливают в стакан тонкой струей воду, предварительно нагретую до 60 – 65 °С. (Соблюдать осторожность – первые порции воды могут вскипеть и попасть в глаза, на кожу!). Когда температура упадет до 100 – 105 о С вводят остальное количество воды из расчета получения колера плотностью 1,35 (при температуре 20 °С), примерно 50% воды к массе сахара. Когда колер охладится до 60–65°С, перемешивание прекращают и колер переливают в колбу.
Для определения плотности полученного колера наливают его в цилиндр на 2/3 его высоты и определяют плотность раствора с помощью ареометра. Полученные по шкале значения записывают для дальнейших расчетов.
Нормально приготовленный колер имеет плотность 1,35 и окрашивающую способность раствора концентрацией 2 г/л, соответствующую по цветомеру высоте столба жидкости 16 ± 2 мм при сравнении с эталоном цвета № 10 и компенсаторе 00; при определении по фотоэлектрокалориметру — оптическую плотность 0,280–0,340 нм при λ = 413 нм и ширине кюветы 3 мм.
Обработка результатов. Выход колера плотностью 1,35 (при температуре 20 °С) составляет 105–108% к массе взятого сахара. Если плотность колера отличается от указанной, то выход его определяют при нормальной плотности с помощью диаграммы Приложения 5.
Например, для варки взято 100 кг сахара-песка. Получено 103,50 кг колера плотностью 1,365. Согласно диаграмме 100 кг колера данной плотности соответствует 103,78 кг колера с нормальной плотностью. Выход колера равен: (103,5 х 103,78)/ 100 = 107,41%
Контрольные вопросы:
1. Цель использования красителей в производстве безалкогольных напитков.
2. Какие красители относятся к натуральным, а какие к синтетическим?
3. Какие химические процессы протекают при получении колера?
Приложение 1
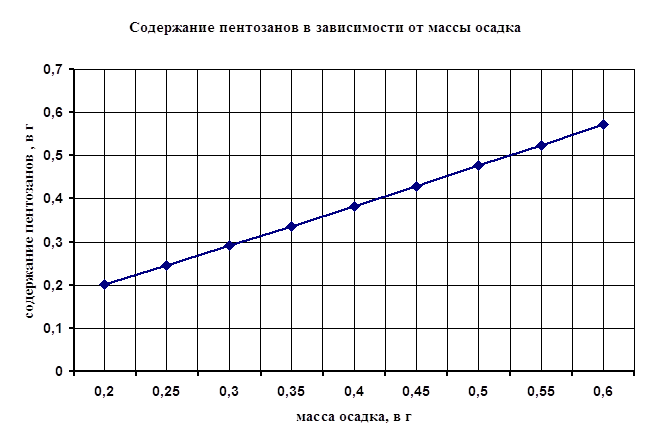
Приложение 2
Таблица 1
Вычисление массовой доли спирта
|
Относите-льная плот- ность |
Массовая доля спирта, % |
Относите-льная плот- ность |
Массовая доля спирта, % |
Относите-льная плот- ность |
Массовая доля спирта, % |
Относите-льная плот- ность |
Массовая доля спирта, % |
|
| 1,000 | 0,000 | 0,9967 | 1,785 | 0,9934 | 3,670 | 0,9901 | 5,700 | |
| 0,9999 | 0,055 | 6 | 1,840 | 3 | 3,730 | 0 | 5,760 | |
| 8 | 0,110 | 5 | 1,890 | 2 | 3,785 | 0,9899 | 5,820 | |
| 7 | 0,165 | 4 | 1,950 | 1 | 3,845 | 8 | 5,890 | |
| 6 | 0,220 | 3 | 2,005 | 0 | 3,905 | 7 | 5,950 | |
| 5 | 0,270 | 2 | 2,060 | 0,9929 | 3,965 | 6 | 6,015 | |
| 4 | 0,325 | 1 | 2,120 | 8 | 4,030 | 5 | 6,080 | |
| 3 | 0,380 | 0 | 2,170 | 7 | 4,090 | 4 | 6,150 | |
| 2 | 0,435 | 0,9959 | 2,225 | 6 | 4,150 | 3 | 6,205 | |
| 1 | 0,485 | 8 | 2,280 | 5 | 4,215 | 2 | 6,270 | |
| 0 | 0,540 | 7 | 2,335 | 4 | 4,275 | 1 | 6,330 | |
| 0,9989 | 0,590 | 6 | 2,390 | 3 | 4,335 | 0 | 6,395 | |
| 8 | 0,645 | 5 | 2,450 | 2 | 4,400 | 0,9889 | 6,455 | |
| 7 | 0,700 | 4 | 2,505 | 1 | 4,460 | 8 | 6,520 | |
| 6 | 0,750 | 3 | 2,560 | 0 | 4,520 | 7 | 6,580 | |
| 5 | 0,805 | 2 | 2,620 | 0,9919 | 4,580 | 6 | 6,645 | |
| 4 | 0,855 | 1 | 2,675 | 8 | 4,640 | 5 | 6,710 | |
| 3 | 0,910 | 0 | 2,730 | 7 | 4,700 | 4 | 6.780 | |
| 2 | 0,960 | 0,9949 | 2,790 | 6 | 4,760 | 3 | 6,840 | |
| 1 | 1,015 | 8 | 2,850 | 5 | 4,825 | 2 | 6,910 | |
| 0 | 1,070 | 7 | 2,910 | 4 | 4,885 | 1 | 6,980 | |
| 0,9979 | 1,125 | 6 | 2,970 | 3 | 4,945 | 0 | 7,050 | |
| 8 | 1,180 | 5 | 3,030 | 2 | 5,005 | 0,9879 | 7,115 | |
| 7 | 1,235 | 4 | 3,090 | 1 | 5,070 | 8 | 7,180 | |
| 6 | 1,285 | 3 | 3,150 | 0 | 5,130 | 7 | 7,250 | |
| 5 | 1,345 | 2 | 3,205 | 0,9909 | 5,190 | 6 | 7,310 | |
| 4 | 1,400 | 1 | 3,265 | 8 | 5,255 | 5 | 7,380 | |
| 3 | 1,455 | 0 | 3,320 | 7 | 5,315 | 4 | 7,445 | |
| 2 | 1,510 | 0,9939 | 3,375 | 6 | 5,375 | 3 | 7,510 | |
| 1 | 1,565 | 8 | 3,435 | 5 | 5,445 | 2 | 7,580 | |
| 0 | 1,620 | 7 | 3,490 | 4 | 5,510 | 1 | 7,650 | |
| 0,9969 | 1,675 | 6 | 3,550 | 3 | 5,570 | 0 | 7,710 | |
| 8 | 1,730 | 5 | 3,610 | 2 | 5,635 |
Таблица 2
Вычисление массовой доли действительного экстракта
|
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
|
||||||||||
| 1,0040 | 1,026 | 1,0086 | 2,203 | 1,0132 | 3,371 | 1,0178 | 4,529 |
|
||||||||||
| 1 | 1,052 | 7 | 2,229 | 3 | 3,396 | 9 | 4,555 |
|
||||||||||
| 2 | 1,078 | 8 | 2,254 | 4 | 3,421 | 1,0180 | 4,580 |
|
||||||||||
| 3 | 1,103 | 9 | 2,280 | 5 | 3,447 | 1 | 4,605 |
|
||||||||||
| 4 | 1,129 | 1,0090 | 2,305 | 6 | 3,472 | 2 | 4,630 |
|
||||||||||
| 5 | 1,155 | 1 | 2,330 | 7 | 3,497 | 3 | 4,655 |
|
||||||||||
| 6 | 1,180 | 2 | 2,356 | 8 | 3,523 | 4 | 4,680 |
|
||||||||||
| 7 | 1,206 | 3 | 2,381 | 9 | 3,548 | 5 | 4,705 |
|
||||||||||
| 8 | 1,232 | 4 | 2,407 | 1,0140 | 3,573 | 6 | 4,730 |
|
||||||||||
| 9 | 1,257 | 5 | 2,432 | 1 | 3,598 | 7 | 4,755 |
|
||||||||||
| 1,0050 | 1,283 | 6 | 2.458 | 2 | 3,624 | 8 | 4,780 |
|
||||||||||
| 1 | 1,308 | 7 | 2,483 | 3 | 3,649 | 9 | 4,805 |
|
||||||||||
| 2 | 1,334 | 8 | 2,508 | 4 | 3,674 | 1,0190 | 4,830 |
|
||||||||||
| 3 | 1,360 | 9 | 2,534 | 5 | 3,699 | 1 | 4,855 |
|
||||||||||
| 4 | 1,385 | 1,0100 | 2,560 | 6 | 3,725 | 2 | 4,880 |
|
||||||||||
| 5 | 1,411 | 1 | 2,585 | 7 | 3,750 | 3 | 4,905 |
|
||||||||||
| 6 | 1,437 | 2 | 2,610 | 8 | 3,775 | 4 | 4,930 |
|
||||||||||
| 7 | 1,462 | 3 | 2,636 | 9 | 3,800 | 5 | 4,955 |
|
||||||||||
| 8 | 1,488 | 4 | 2,661 | 1,0150 | 3,826 | 6 | 4,980 |
|
||||||||||
| 9 | 1,514 | 5 | 2,687 | 1 | 3,851 | 7 | 5,005 |
|
||||||||||
| 1,0060 | 1,539 | 6 | 2,712 | 2 | 3,876 | 8 | 5,030 |
|
||||||||||
| 1 | 1,565 | 7 | 2,738 | 3 | 3,901 | 9 | 5,055 |
|
||||||||||
| 2 | 1,590 | 8 | 2,763 | 4 | 3,926 | 1,0200 | 5,080 |
|
||||||||||
| 3 | 1,616 | 9 | 2,788 | 5 | 3,951 | 1 | 5,106 |
|
||||||||||
| 4 | 1,641 | 1,0110 | 2,814 | 6 | 3,977 | 2 | 5,130 |
|
||||||||||
| 5 | 1,667 | 1 | 2,839 | 7 | 4,002 |
3 J |
5,155 |
|
||||||||||
| 6 | 1,693 | 2 | 2,8.64 | 8 | 4,027 | 4 | 5,180 |
|
||||||||||
| 7 | 1,718 | 3 | 2,890 | 9 | 4,052 | 5 | 5,205 |
|
||||||||||
| 8 | 1,744 | 4 | 2,915 | 1,0160 | 4,077 | 6 | 5,230 |
|
||||||||||
| 9 | 1,769 | 5 | 2,940 | 1 | 4,102 | 7 | 5,255 |
|
||||||||||
| 1,0070 | 1,795 | 6 | 2,966 | 2 | 4,128 | 8 | 5,280 |
|
||||||||||
| 1 | 1,820 | 7 | 2,991 | 3 | 4,153 | 9 | 5,305 |
|
||||||||||
| 2 | 1,846 | 8 | 3,017 | 4 | 4,178 | 1,0210 | 5,330 |
|
||||||||||
| 3 | 1,872 | 9 | 3,042 | 5 | 4,203 | 1 | 5,355 |
|
||||||||||
| 4 | 1,897 | 0,0120 | 3,067 | 6 | 4,228 | 2 | 5,380 |
|
||||||||||
| 5 | 1,923 | 1 | 3,093 | 7 | 4,253 | 3 | 5,405 |
|
||||||||||
| 6 | 1,948 | 2 | 3,118 | 8 | 4,278 | 4 | 5,430 |
|
||||||||||
| 7 | 1,973 | 3 | 3,143 | 9 | 4,304 | 5 | 5,455 |
|
||||||||||
| 8 | 1,999 | 4 | 3,169 | 1,0170 | 4,329 | 6 | 5,480 |
|
||||||||||
| 9 | 2,025 | 5 | 3,194 | 1 | 4,354 | 7 | 5,505 |
|
||||||||||
| 1,0080 | 2,053 | 6 | 3,219 | 2 | 4,379 | 8 | 5,530 |
|
||||||||||
| 1 | 2,078 | 7 | 3,245 | 3 | 4,404 | 9 | 5,555 |
|
||||||||||
| 2 | 2,101 | 8 | 3,270 | 4 | 4,429 | 1,0220 | 5,580 |
|
||||||||||
| 3 | 2,127 | 9 | 3,295 | 5 | 4,454 | 1 | 5,605 |
|
||||||||||
| 4 | 2,152 | 1,0130 | 3,321 | 6 | 4,479 | 2 | 5,629 |
|
||||||||||
| 5 | 2,178 | 1 | 3,346 | 7 | 4,505 | 3 | 5,654 |
|
||||||||||
|
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
|||||||||||
| 1,0224 | 5,679 | 1,0271 | 6,844 | 1,0318 | 8,000 | 1,0365 | 9,145 | |||||||||||
| 5 | 5,704 | 2 | 6,868 | 9 | 8,024 | 6 | 9,170 | |||||||||||
| 6 | 5.729 | 3 | 6,893 | 1.0320 | 8,048 | 7 | 9,194 | |||||||||||
| 7 | 5,754 | 4 | 6,918 | 1 | 8,073 | 8 | 9,218 | |||||||||||
| 8 | 5,779 | 5 | 6,943 | 2 | 8.098 | 9 | 9.243 | |||||||||||
| 9 | 5,803 | 6 | 6,967 | 3 | 8,122 | 1,0370 | 9,267 | |||||||||||
| 1,0230 | 5,828 | 7 | 6,992 | 4 | 8,146 | 1 | 9,291 | |||||||||||
| 1 | 5,853 | 8 | 7,017 | 5 | 8,171 | 2 | 9,316 | |||||||||||
| 2 | 5,878 | 9 | 7,041 | 6 | 8,195 | 3 | 9,340 | |||||||||||
| 3 | 5,903 | 1,0280 | 7,066 | 7 | 8,220 | 4 | 9,364 | |||||||||||
| 4 | 5,928 | 1 | 7,091 | 8 | 8,244 | 5 | 9,388 | |||||||||||
| 5 | 5.952 | 2 | 7,113 | 9 | 8.269 | 6 | 9,413 | |||||||||||
| 6 | 5,977 | 3 | 7,140 | 1,0330 | 8,293 | 7 | 9,437 | |||||||||||
| 7 | 6,002 | 4 | 7,164 | 1 | 8,317 | 8 | 9,461 | |||||||||||
| 8 | 6,027 | 5 | 7,189 | 2 | 8.342 | 9 | 9,485 | |||||||||||
| 9 | 6,052 | 6 | 7,214 | 3 | 8,366 | 1,0380 | 9,509 | |||||||||||
| 1,0240 | 6,077 | 7 | 7,238 | 4 | 8,391 | 1 | 9,534 | |||||||||||
| 1 | 6,101 | 8 | 7,263 | 5 | 8,415 | 2 | 9,558 | |||||||||||
| 2 | 6,126 | 9 | 7,287 | 6 | 8,439 | 3 | 9,582 | |||||||||||
| 3 | 6,151 | 1,0290 | 7,312 | 7 | 8,464 | 4 | 9,606 | |||||||||||
| 4 | 6,176 | 1 | 7,337 | 8 | 8,488 | 5 | 9,631 | |||||||||||
| 5 | 6,200 | 2 | 7,361 | 9 | 8,513 | 6 | 9,655 | |||||||||||
| 6 | 6,225 | 3 | 7,386 | 1,0340 | 8,537 | 7 | 9,679 | |||||||||||
| 7 | 6,250 | 4 | 7,411 | 1 | 8,561 | 8 | 9,703 | |||||||||||
| 8 | 6,275 | 5 | 7,435 | 2 | 8,586 | 9 | 9,727 | |||||||||||
| 9 | 6,300 | 6 | 7,460 | 3 | 8.610 | 1,0390 | 9,751 | |||||||||||
| 1,0250 | 6,325 | 7 | 7,484 | 4 | 8,634 | 1 | 9,776 | |||||||||||
| 1 | 6,350 | 8 | 7,509 | 5 | 8,659 | 2 | 9,800 | |||||||||||
| 2 | 6,374 | 9 | 7,533 | 6 | 8,683 | 3 | 9,824 | |||||||||||
| 3 | 6,399 | 1,0300 | 7,558 | 7 | 8,708 | 4 | 9,848 | |||||||||||
| 4 | 6,424 | 1 | 7,583 | 8 | 8,732 | 5 | 9,873 | |||||||||||
| 5 | 6,449 | 2 | 7,607 | 9 | 8,756 | 6 | 9,897 | |||||||||||
| 6 | 6,473 | 3 | 7,632 | 1,0350 | 8,781 | 7 | 9,921 | |||||||||||
| 7 | 6,498 | 4 | 7,656 | 1 | 8,805 | 8 | 9,945 | |||||||||||
| 8 | 6,523 | 5 | 7,681 | 2 | 8,830 | 9 | 9,969 | |||||||||||
| 9 | 6,547 | 6 | 7,705 | 3 | 8,854 | 1,0400 | 9,993 | |||||||||||
| 1,0260 | 6,572 | 7 | 7,730 | 4 | 8,878 | 1 | 10,017 | |||||||||||
| 1 | 6,597 | 8 | 7,754 | 5 | 8,902 | 2 | 10,042 | |||||||||||
| 2 | 6,621 | 9 | 7,779 | 6 | 8,927 | 3 | 10,066 | |||||||||||
| 3 | 6,646 | 1,0310 | 7,803 | 7 | 8,951 | 4 | 10,090 | |||||||||||
| 4 | 6,671 | 1 | 7,828 | 8 | 8,975 | 5 | 10,114 | |||||||||||
| 5 | 6,696 | 2 | 7,853 | 9 | 9,000 | 6 | 10,138 | |||||||||||
| 6 | 6,720 | 3 | 7,877 | 1,0360 | 9,024 | 7 | 10,162 | |||||||||||
| 7 | 6,745 | 4 | 7,901 | 1 | 9.048 | 8 | 10,186 | |||||||||||
| 8 | 6,770 | 5 | 7,926' | 2 | 9,073 | 9 | 10,210 | |||||||||||
| 9 | 6,794 | 6 | 7,950 | 3 | 9,087 | 1,0410 | 10,234 | |||||||||||
| 1,0270 | 6,819 | 7 | 7,975 | 4 | 9,121 | 1 | 10,259 | |||||||||||
|
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
Относи- тельная плотность |
Массовая доля действительного экстракта, % |
|
||||||||||
| 1,0412 | 10,283 | 1,0432 | 10,764 | 1,0452 | 11,243 | 1,0472 | 11,721 |
|
||||||||||
| 3 | 10,307 | 3 | 10,788 | 3 | 11,267 | 3 | 11,745 |
|
||||||||||
| 4 | 10,331 | 4 | 10,812 | 4 | 11,291 | 4 | 11,768 |
|
||||||||||
| 5 | 10,355 | 5 | 10,836 | 5 | 11,315 | 5 | 11,792 |
|
||||||||||
| 6 | 10,379 | 6 | 10,860 | 6 | 11,339 | 6 | 11,816 |
|
||||||||||
| 7 | 10,403 | 7 | 10,884 | 7 | 11,363 | 7 | 11,840 |
|
||||||||||
| 8 | 10,427 | 8 | 10,908 | 8 | 11,387 | 8 | 11,864 |
|
||||||||||
| 9 | 10,451 | 9 | 10,932 | 9 | 11,411 | 9 | 11,888 |
|
||||||||||
| 1,0420 | 10,475 | 1,0440 | 10,956 | 1,0460 | 11,435 | 1,0480 | 11,912 |
|
||||||||||
| 1 | 10,499 | 1 | 10,980 | 1 | 11,458 | 1 | 11,935 |
|
||||||||||
| 2 | 10,523 | 2 | 11,004 | 2 | 11,482 | 2 | 11,959 |
|
||||||||||
| 3 | 10,548 | 3 | 11,027 | 3 | 11,506 | 3 | 11,983 |
|
||||||||||
| 4 | 10,571 | 4 | 11,051 | 4 | 11,530 | 4 | 12,007 |
|
||||||||||
| 5 | 10,569 | 5 | 11,075 | 5 | 11,554 | 5 | 12,031 |
|
||||||||||
| 6 | 10,620 | 6 | 11,100 | 6 | 11,578 | 6 | 12,054 |
|
||||||||||
| 7 | 10,644 | 7 | 11.123 | 7 | 11,602 | 7 | 12,078 |
|
||||||||||
| 8 | 10,668 | 8 | 11,147 | 8 | 11,626 | 8 | 12,102 |
|
||||||||||
| 9 | 10,692 | 9 | 11,171 | 9 | 11,650 | 9 | 12,126 |
|
||||||||||
| 1,0430 | 10,716 | 1,0450 | 11,195 | 1,0470 | 11,673 | 1,0490 | 12,150 |
|
||||||||||
| 1 | 10,740 | 1 | 11,219 | 1 | 11,697 |
|
||||||||||||
Приложение 3
Составление расчетного уравнения. Уравнение прямой, полученной на градуировочном графике (см. п. 7.3), имеет вид Х = ау+b,
т.е. в данном случае для оптической плотности D: Cал = aD + b
Для определения неизвестных параметров а и b следует составить четыре уравнения для четырех значений Сал и соответствующих им значений оптических плотностей D.
Например, после колориметрирования анализируемых растворов с содержанием альдегидов 2; 3; 4 и 10 (мг/дм3 безводного спирта) получены значения оптических плотностей, равные соответственно 0,185; 0,260; 0,335 и 0,810.
Подставляя полученные значения в уравнение Cал = aD + b, получаем четыре уравнения, которые объединяем по два и складываем:
|
2 = 0,185 а + b + 3 = 0,260 а + b 5 = 0,445 а+ 2b |
4 = 0,335 a + b + 10 = 0,810 a + b 14 = 1,145 a + 2b |
Определяем коэффициенты a и b:
14 = 1,145a + 2b
– 5 = 0,445 а + 2b
9 = 0,700 а; а= 12,71.
Подставляя значение а в одно из уравнений, находим значение b:
5 = 0,445-12,71+2 b; b = - 0,32.
Подставляя полученные значения коэффициентов a и b получаем расчетное уравнение Сал = 12,71D - 0,32, где D – оптическая плотность.
Полученное уравнение используют для вычисления массовой концентрации альдегидов в водках.
Приложение 4
Физико–химические показатели водок
Показатели |
Норма для обыкновенных водок из спирта | Норма для водок особых из спирта | ||||
| высшей очистки | Экстра (за исключением Посольской) |
Экстра для Посольской |
Люкс | Экстра | высшей очистки | |
|
Крепость, % Щелочность (объем соляной кислоты концентрацией 0,1 моль/дм3, израсходованной на титрование 100 см3 водки), см3, не более Массовая концентрация, мг, не более: альдегидов в пересчете на уксусный альдегид в 1 дм3 безводного спирта сивушного масла в пересчете на смесь изоамилового и изобутилового спиртов (3:1) в 1 дм3 безводного спирта эфиров в пересчете на уксусно-этиловый эфир в 1 дм3 безводного спирта Объемная доля метилового спирта в пересчете на безводный спирт, %, не более |
40–45, 50,56 3,5 8 4 30 0,05 |
38–45, 50,56 3,0 3 3 25 0,03 |
40 3,5 6 4 25 0,03 |
40 3,5 3 2 18 0,03 |
40 3,0 3 3 25 0,03 |
40-45 3,5 8 4 30 0,05 |
Приложение 5
Список литературы
1. Полыгалина Г.В., Технохимический контроль спиртового и ликероводочного производств.-М: Колос,1999. –336 с..
2. Методы технохимического контроля в виноделии. Под ред. Гержиновой В.Г.-Симферополь: «Таврида», 2002. -260 с.
3. Хорунжина С.И. Биохимические и физико–химические основы технологии солода и пива.-М: Колос, 1999. –312 с..
4. Косминский Г.И. Технология солода, пива и безалкогольных напитков. Лабораторный практикум по технохимическому контролю производства. – Мн.: Дизайн ПРО, 1998. – 352 с.
5. Кишковский З.Н., Скурихин И.М. Химия вина. -М.:Пищевая промышленность,1976.–253с.
6. Нилов В. И., Скурихин И. М. Химия виноделия. –М.: 1967. –442 с.
7. Калунянц К.А. Химия солода и пива. –М: Агропромиздат,1990.
| Экспертиза качества пива | |
|
... изделия 1.1 Потребительские свойства изделия 1.2 Технологический процесс как фактор, влияющий на формирование качества пива 1.3 Требования к качеству Кислотность пива раствора гидроокиси натрия концентрацией 1 моль/дм3 на 100 см3 пива вычисляют по формуле Остаток после отгонки спирта доводят в колбе дистиллированной водой до первоначальной массы пива 100 см3, перемешивают, определяют плотность пикнометром при температуре (20,0=0,2 ... |
Раздел: Рефераты по кулинарии Тип: курсовая работа |
| Развитие, становление и основные аспекты фармации | |
|
РАЗВИТИЕ, СТАНОВЛЕНИЕ И ОСНОВНЫЕ АСПЕКТЫ ФАРМАЦИИ Для ветеринарного провизора необходимы знания, с помощью которых можно контролировать качество ... Тартраты обнаруживают по связанной винной кислоте, осаждая ее ионом калия, салицилаты и бензоаты открывают ионом железа (III), лактаты испытывают с помощью реакции на молочную ... испытание на соли железа (II) и (III) в зависимости от концентрации основано на образовании с раствором сульфосалициловой кислоты в аммиачной среде коричнево-красных или желтых ... |
Раздел: Рефераты по медицине Тип: книга |
| Технология производства и товароведная оценка светлых сортов пива | |
|
Реферат Данная работа содержит _____ страниц, _ иллюстраций, _____таблиц, использовано _____источника литературы. Перечень ключевых слов: затирание ... Пиво представляет собой водно-спиртовой раствор слабой концентрации, поэтому при умеренном употреблении пива спирт сжигается в организме человека до CO2 и H2O почти без остатка. Зола пива представлена солями натрия и калия (приблизительно 30%), солями фосфорной кислоты (приблизительно 30%), кремневой кислоты (около10%), небольшим количеством Ca, Mg, Al, Fe ... |
Раздел: Рефераты по технологии Тип: реферат |
| Проблемы водоснабжения России | |
|
КУРСОВАЯ РАБОТА на тему: "Проблемы водоснабжения России" Оглавление Введение Глава I Литературный обзор 1.1 Основные типы вод 1.2 Формирование ... В коническую колбу вместимостью 250 см3 наливают 70-80 см3 дистиллированной воды, добавляют пипеткой 10 см3 раствора дихромата калия с концентрацией 0,0200 моль/дм3 эквивалента ... Оптическую плотность растворов с концентрацией сероводорода 5-30 мкг/дм3 измеряют в кюветах с толщиной слоя 2 см, оптическую плотность растворов с концентрацией 20-80 мкг/дм3 - в ... |
Раздел: Рефераты по экологии Тип: дипломная работа |
| История и производство пива | |
|
Тема История и производство пива 1. История развития (Всемирная история пива) 1.1 Общая история Забывчивый пекарь, когда то еще в такой далекой от нас ... 4.1.5 Массовая концентрация дрожжевых клеток в неосветленном пиве не более 2 млн.кл/см3. см3 0,1 моль/дм3 раствора йода на 100см3 воды |
Раздел: Рефераты по кулинарии Тип: курсовая работа |