Реферат: Процеси окиснення вуглеводнів
Процеси окиснення вуглеводнів
Зміст
Вступ
1. Характеристика процесів окиснення. Визначення і класифікація реакцій окиснення
3.1 Гідропероксиди
3.2 Карбонові кислоти
3.3 Альдегіди
3.4 Кінетика і каталіз реакції радикально-ланцюгового окиснення
3.5 Селективність окиснення
4.1 Характеристика реакторів
5. Окиснення вуглеводнів у гідро пероксиди
7.1 Теоретичні основи процесу
7.1.1 Гетерогенні каталізатори окиснення і механізм реакцій
7.1.2 Кінетика і селективність гетерогенно-каталітичного окиснення
7.2 Реактори процесів гетерогенно-каталітичного окиснення
7.3 Окиснення олефінів по насиченому атому вуглецю
Література
Вступ
Тема реферату «Процеси окиснення вуглеводнів» з дисципліни «Технологія нафтохімічного синтезу».
Практичне значення процесів окиснення у виробництві основного органічного та нафтохімічного синтезу важко переоцінити. Їх першорядну роль обумовили наступні причини:
1.Велика цінність сполук, отриманих окисненням (спиртів, альдегідів, кетонів, карбонових кислот та їх ангідридів, a-оксидів, нітрилів та ін.) і проміжними продуктами органічного синтезу, розчинниками, мономерами і вихідними речовинами для виробництва полімерних матеріалів, пластифікаторів і т.д.
2.Широке різноманіття реакцій окиснення, до яких здатні багато органічних речовин, у тому числі вуглеводні всіх класів. Це дозволяє використовувати процеси окиснення для первинної переробки вуглеводневої сировини і робити на їх основі велику кількість коштовних речовин.
3.Приступність і низька вартість більшості окиснювачів, серед яких головне місце займає кисень повітря. Це визначає більш високу економічність синтезу деяких продуктів методами окиснення в порівнянні з іншими можливими методами їх виробництва.
Окисні процеси одержали широке поширення в органічному синтезі, часто заміняючи менш економічні способи виробництва багатьох продуктів.
1. Характеристика процес ів окиснення. Визначення і класифікація реакцій окиснення
В органічній хімії і технології процесами окиснення вважаються перетворення речовин під дією тих або інших окиснювачів. Існує повне або неповне окиснення. Під першим розуміють згорання речовин з утворенням діоксиду вуглецю і води:
С3Н8 + 5О2 ® 3СО2 + 4Н2О
В органічному синтезі повне окиснення є небажаним побічним процесом. Для синтезу важливі лише реакції неповного окиснення, які можна розділити на три важливі групи:
1.Окиснення без розриву вуглецевого зв'язку, коли число атомів вуглецю залишається таким же, як і у вихідному з'єднанні.
Ці реакції поділяються на дві групи:
- окиснення по насиченому атому вуглецю в парафінах, нафтенах, олефінах, алкілароматичних вуглеводнях і в похідних цих з'єднань, особливо в спиртах і альдегідах:
+0,5О2+0,5О2
 СН3-СН2-СН(ОН)-СН3
СН3-СН2-СН(ОН)-СН3
СН3-СН2-СН2-СН3 ® О2
СН3-СН2-СО-СН3 + Н2О
СН2=СН-СН3 ![]() СН2=СН-СНО +
Н2О
СН2=СН-СНО +
Н2О
С6Н5СН3 ![]() С6Н5СНО
С6Н5СНО ![]() С6Н5СООН
С6Н5СООН
- окиснення за подвійним зв'язком з утворенням a-оксидів (епоксидування) карбонільних з'єднань або гликолів:
![]()
СН2=СН2 + 0,5О2 ® СН2-СН2О
RCH=CH2 + 0,5О2 ® RCOCH3
RCH=CH2 + H2O2 ® RCHOH-CH2OH
2.Деструктивне окиснення, що протікає з розщепленням вуглець-вуглецевих зв'язків. До нього здатні вуглеводні (та їх похідні) ряду парафінів, нафтенів, олефінів і ароматичні вуглеводні. Деструкція протікає по зв'язках С-С, С=С або Сар-Сар:
СН3-СН2-СН2-СН3 ![]() 2СН3-СООН
+ Н2О
2СН3-СООН
+ Н2О
RCH=CHR¢![]() RCOOH + R¢COOH
RCOOH + R¢COOH

![]() HOOC-(CH2)4-COOH
+ H2O
HOOC-(CH2)4-COOH
+ H2O
CO
C
![]() êêO + 2CO2 + 2H2O
êêO + 2CO2 + 2H2O
C
CO
3.Окиснення, що супроводжується зв'язуванням молекул вихідних реагентів (окисна конденсація або окисне сполучення):
О
СН2=СН2 + СН3-СООН + 0,5О2![]() СН2=СН-О-С-СН3
СН2=СН-О-С-СН3
ОО
СН2=СН2 + 2СН3-СООН + 0,5О2![]() СН3-С-О-СН2 –
СН3-С-О-СН2 –
СН2-О-С-СН3
2RSH ![]() RSSR + H2O
RSSR + H2O
2RH ![]() ROOR + H2O
ROOR + H2O
RCH3 + NH3 ![]() RCN +3H2O
RCN +3H2O
(окиснювальний амоноліз)
2.Окиснювальні агенти і техн іка безпеки у процесах окиснення
У промисловості основного органічного синтезу використовуються дешеві окиснювачі і лише в окремих випадках застосовують агенти, здатні до реакцій, що не протікають у присутності інших окиснювачів.
2.2 Молекулярний кисень
Найважливіший окисний агент використовується у виді повітря, технічного кисню, азотнокислих сумішей з невеликим вмістом кисню. Концентрований кисень робить більш сильну окиснюючу дію, але його застосування зв'язане з додатковими витратами на поділ повітря. При окисненні в газовій фазі, коли суміш азоту утрудняє виділення продуктів або їх рециркуляцію, використовують і технічний кисень. Меншу швидкість реакції при окисненні повітрям компенсують підвищенням температури або збільшенням загального тиску, що веде до росту парціального тиску кисню.
2.3 Азотна кислота
Азотна кислота (рідше оксиди азоту) служить другим за масштабами застосування окисним агентом. Її дія нерідко супроводжується побічним нитруванням органічної сполуки, що підсилюється з підвищенням концентрації кислоти. З цієї причини для окиснення використовують 40-60 %-ву азотну кислоту. Азотна кислота як окиснювач ніколи не застосовується для реакцій з парафінами. Для неї найбільш типовими реакціями є реакції деструктивного окиснення циклічних з'єднань і речовин з ненасиченими зв'язками, що йдуть за участю азотної кислоти, із кращим виходом, чим при окиснюванні киснем.
![]()
—ОН ![]() НООС-(СН2)4-СООН + 2N2O3 + 3H2O
НООС-(СН2)4-СООН + 2N2O3 + 3H2O
При окисненні азотною кислотою вона розкисняється до оксидів азоту (NO або N2O3). Економічність виробництва багато в чому залежить від можливості утилізації цих оксидів і регенерації неперетвореної азотної кислоти. Перша задача вирішується окиснюванням оксидів азоту повітрям у водному або азотнокислому розчині з утворенням азотної кислоти:
N2O3 + O2 + H2O ® 2HNO3
2.4 Пероксидні з’єднання
Пероксидні з'єднання, головним чином пероксид водню і перуксусна кислота (а останнім часом і гидропероксиди), набутили застосування як окисні агенти в основному органічному та нафтохімічному синтезі порівняно недавно. Через відносну дорожнечу їх використовують тільки для таких реакцій, що не протікають під впливом молекулярного кисню або азотної кислоти. Це відноситься насамперед до процесів епоксидирування ненасичених з'єднань:
![]()
-ООН + RCH=CH2 ® -OH + RCH-CH2O
Пероксид водню звичайно застосовують у виді 30 %-вого водного розчину. Він дає з карбоновими кислотами відповідні пероксикислоти за реакціэю, аналогічною реакції етерифікації:
RСООН + Н2О2 « RСОООН + Н2О
Пероксикислоти утворюються, крім того, окисненням альдегідів. Так, перуксусна кислота виробляється цим шляхом у промисловому масштабі, а гідропероксиди одержують у промисловості окиснюванням вуглеводнів:
СН3-СНО + О2 ® СН3-СОООН
С6Н5-СН(СН3)2 + О2 ® С6Н5-С(СН3)2
ООН
2.5 Техніка безпеки процесів окиснення
Окисні агенти дають з органічними речовинами вибухонебезпечні суміші або є з'єднаннями, схильними до розкладання. Вибухонебезпечні властивості газоподібних сумішей вуглеводнів з повітрям і дані про температури спалаху рідких вуглеводнів наведені в спеціальній довідковій літературі.
Азотна кислота та інші окиснювачі також дають вибухонебезпечні суміші з органічними речовинами. Небезпека присутності пероксиду водню і перуксусної кислоти посилюється з тієї причини, що реакції їх розпаду з виділенням відповідно води та оцтової кислоти є екзотермічними.
Розкладання пероксидних з'єднань каталізується деякими металами перемінної валентності (залізо, мідь, марганець, кобальт, хром) та їх солями. Тому концентрований пероксид водню та особливо пероксикислоти здатні вибухати під час відсутності органічних речовин. Застосування їх у розчинах і при контрольованому температурному режимі дозволяє уникнути цих утруднень.
2.6 Е нергетична характеристика реакцій окиснення
Усі реакції окиснення, які знайшли застосування у промисловості основного органічного синтезу, необоротні. Це не означає, що їх узагалі не можна провести в зворотному напрямку (наприклад, відновити кислоти в альдегіди, а карбонільні з'єднання в спирти і вуглеводні), але для здійснення зворотної реакції потрібні дії відновлювачей або водню. Отже, окислення практично необоротне, тому що його кінцеві продукти – СО2 і Н2О – не можуть служити відновлювачами.
Окиснення – екзотермічний процес. Найбільше екзотермічними є процеси утворення карбонових кислот, менш - деструктивне окиснення парафінів і ще менше – ароматичних систем (відповідно -DН0298 = 567,4; 982-1003; 1807 кДж/моль).
Менш екзотермічними процесами є процеси утворення карбонільних з’єднань з вуглеводнів (-DН0298 = 355; 284; 218 кДж/моль), карбонових кислот з альдегідів (DН0298 = 260 кдж/моль). Тепловий ефект є меньшим при утворенні спиртів, a-оксидів.
3. Радикально-ланцюгове окиснення
Радикально-ланцюгове окислювання включає три групи:
1.Окиснення парафінів та їх похідних.
2.Окиснення циклопарафінів та їх похідних.
3.Окиснення бічних ланцюгів алкілароматичних вуглеводнів.
При окисненні вуглеводнів утворюється цілий ряд молекулярних продуктів: гідропероксиди, спирти, кетони, альдегіди, карбонові кислоти, складні ефіри і деякі, більш складні поліфункціональні з'єднання. Проміжними активними частками є радикали з вільною валентністю на атомі вуглецю (R·) або на кисневих атомах (ROO·, RCOOO·).
3.1 Гідропероксиди
Гідропероксиди – це первинні молекулярні продукти окиснення вуглеводнів. Ланка ланцюга при їх утворенні така:
R· + О2 ® ROO·,
ROO· + RH ® ROOH + R·
Гідропероксиди відносяться до числа досить нестабільних з'єднань, що перетворюються при окисненні в інші продукти. Тому їх концентрація в реакційній масі, особливо при каталітичному окисненні і при підвищених температурах, невелика. Найбільш нестабільні первинні гідропероксиди (RCH2OOН, ArCH2OOH) і навпаки, відносно стабільні третинні гідропероксиди, з яких гідропероксиди ізобутану (СН3)3СООН і ізопропілбензолу С6Н5С(СН3)2ООН є промисловими продуктами. З вторинних відносно стабільні гідропероксиди циклоалканів С8-С12, олефінів і алкілароматичних з'єднань.
Спирти і карбонільні з'єднання є вторинними продуктами окиснення вуглеводнів. Спирти виходять тільки при окисненні парафінів і нафтенів:
спирт
|
 Алкан
Алкан кетон
Гідропероксиди при розкладанні під дією підвищеної температури або каталізаторів окиснення дають спирти і карбонільні з'єднання. При одержанні спиртів ланка ланцюгу така:
ROOH + R· ® ROH + RO·,
RO· + RH ® ROH + R·
Кетони утворюються з вторинних гідропероксидів через стадію радикал-гідропероксидів:
R2CHOOH
+ HO·![]() R2
R2![]() OOH ® R2C=O + HO·
OOH ® R2C=O + HO·
Третинні гідропероксиди при ланцюговому перетворенні дають крім спирту, з тим же числом вуглецевих атомів, також спирт і кетон, але з меншим числом атомів вуглецю за рахунок деструкції вуглець-вуглецевого зв'язку:
R3COOH + R· ® ROH + R3CO·
R3CO· ® R2C=O + R·
Ці механізми реакції характерні для некаталітичного окиснення в рідкій фазі при помірних температурах. При високотемпературному окисненні в газовій фазі всі продукти утворюються через пероксидні радикали, минаючи гідропероксиди, причому відбувається значна деструкція за вуглець-вуглецевим зв'язком з утворенням спиртів і альдегідів:
СН3-СН2-СН2ОО· ® НСНО + СН3-СН2О· ![]() НСНО +
НСНО +
СН3-СН2ОН
СН3-СН-СН3 ® СН3СНОСН3О· ![]() СН3-СНО + СН3ОН
СН3-СНО + СН3ОН
ОО·
Зараз вважають, що при рідинофазному окисненні утворення продуктів відбувається через пероксидні радикали:
R2CH ® R2C· ® R2C=O + HO·OO· OOH
При каталізі солями металів перемінної валентності останні можуть давати комплекси з пероксидними радикалами, що перетворюються в координаційній сфері центрального іона, окисняючи його у вищий валентний стан:
![]() + Co(OAc)2 «
+ Co(OAc)2 « ![]() Co(OAc)2 ® R2C=O + HOCo(OAc)2
Co(OAc)2 ® R2C=O + HOCo(OAc)2
Таким чином, спирти і карбонільні з'єднання можуть виходити при рідинофазному окисненні не тільки послідовно стосовно гідропероксиду, але і паралельно з ним.
3.2 Карбонові кислоти
Карбонові кислоти утворяться при окисненні вуглеводнів зі збереженням їх вуглецевого ланцюгу або з деструкцією за С-С-зв'язком. Перше можливо лише при перетвореннях первинних гідропероксидів і типово тільки лише для окиснення метильных груп алкілароматичних з'єднань через проміжну стадію альдегідів:
ArCH3 ![]() ArCH2OOH
ArCH2OOH ![]() ArCHO
ArCHO ![]() ArCOOH
ArCOOH
При окисненні парафінів і нафтенів карбонові кислоти утворяться з деструкцією вуглецевого ланцюгу. Безпосередніми попередниками кислот є кетони, які окиснюються легше, утворюючи кетопероксидний радикал і кетогідропероксид:
RCH2-COR¢![]() RCH-COR¢
RCH-COR¢![]() RCH-COR¢ OO·OOH
RCH-COR¢ OO·OOH
RCH-COR¢ ® RCHO + RCOOH![]() RCOOH + R¢COOH OOH
RCOOH + R¢COOH OOH
3.3 Альдегіди
Альдегіди є найбільше легко окиснюємимі з'єднаннями, тому при окиснюванні вуглеводнів у рідкій фазі вони або утворяться в невеликій кількості, або їх узагалі не вдається знайти в продуктах реакції. При радикальному окисненні вони дають проміжні ацильний і пероксиацильний радикали і пероксикислоту:
![]() =O + O2 ® RCOOO·
=O + O2 ® RCOOO·
RCOOO· + RCHO ®
RCOOOH + ![]() =O
=O
Крім пероксикислоти і карбонової кислоти
СН3-СОООН + СН3-СНО ® 2СН3-СООН
іншими продуктами окиснення є ангідриди. Їх утворенню сприяє застосування змішаного каталізатору (солі кобальту або марганцю із солями міді) і знижений парціальний тиск кисню.
3.4 Кінетика і каталіз реакції радикально-ланцюгового окиснення
Гомогенне радикально-ланцюгове окиснення складається зі стадій зародження, продовження, обриву і выродженного розгалуження ланцюгу. Первинне утворення радикалів при окисненні відбувається при додаванні в суміш ініціаторів (гідропероксиди і пероксиды, 2,2-азо-біс-ізобутиронітрил в рідкій фазі: HNO3, NO і HBr в газовій фазі), за рахунок автоокиснения органічної речовини або при взаємодії кисню з каталізатором:
RH + O2 ® R· + HOO·
Cо2+ +O2 ® Cо3+-OO·
У розвиненому процесі окиснення здобувають значення й інші джерела радикалів, серед яких важливу роль відводять реакціям виродженного розгалуження ланцюгу. При високотемпературному окиснюванні в газовій фазі воно досягається окиснюванням реакційноздатних альдегідів, а при помірній температурі рідинофазных процесів – шляхом розкладання гідропероксидів або пероксикислот:
RСНО
+ O2 ® ![]() =О + НОО·
=О + НОО·
2RООН ® RО· + RОО· + Н2О
Остання реакція прискорюється солями металів перемінної валентності (ацетати і нафтени кобальту, марганцю та ін.):
RООН + Mn(OAс)2 ® RО· + HOMn(OAс)2
RООOН + Co(OAс)2 ® ROО· + HOCo(OAс)2
Утворення радикалів може відбуватися за рахунок окиснення гідропероксидів, альдегідів або вуглеводнів валентною формою каталізатора:
RООН + Mn3+ ® RОО· + Mn2+ + Н+
RН + Co3+ ® R· + Co2+ + Н+
Стадії продовження ланцюгу та утворення продуктів розглядалися раніше.
Обрив ланцюгу при газофазному окисненні звичайно протікає лінійно при зіткненні пероксидного радикалу зі стінкою:
НОО· ® НООадс
При рідинофазному окисненні відбувається обрив на найменш реакційно-здатних пероксидних або пероксиацильних радикалах з утворенням молекулярних продуктів.
Обрив ланцюгу може відбуватися на інгібіторах (сіркоміски з'єднання, феноли).
На практиці некаталітичне окиснення в рідкій фазі застосовують тільки при синтезі гідропероксидів і пероксикислот:
R· + O2
![]() ROO·
ROO·
ROO· + RH ![]() ROOH +R·
ROOH +R·
2ROOH ![]() RO· + ROO· + H2O
RO· + ROO· + H2O
ROOH ![]() RO· + HO·
RO· + HO·
2ROO· ![]() молекулярные продукты
молекулярные продукты
Для інтенсифікації початкової стадії окиснення вигідно додавати у вихідну суміш готовий гідропероксид.
При каталітичному окисненні, наприклад альдегідів, кінетику процесу визначають такі елементарні стадії:
RСНО
+ Со3+ ® ![]() + Со2+ + Н+
+ Со2+ + Н+
![]() + О2 ® RСОOO·
+ О2 ® RСОOO·
RСОOO· + RСНО ® RСООOН + ![]()
RСООOН + Со2+ ® RСОO· + HO- + Со3+
2RСОOO· ® молекулярні продукти
Енергія активації при гомогенно-каталітичному окисненні вуглеводнів у рідкій фазі складає 50-84 кДж/моль у порівнянні з 105-147 кДж/моль при термічному або ініційованому окисненні.
3.5 Селективність окиснення
Селективність залежить від розвитку рівнобіжних і послідовних перетворень при утворенні цільових і побічних продуктів. Рівнобіжні перетворення можуть бути обумовлені двома факторами: реакціями по різних атомах вуглецю в молекулі вихідного реагенту або рівнобіжним утворенням речовин з різними функціональними групами. Перші реакції залежать від відносної реакційної здатності різних атомів водню при атаці пероксидним або іншим кисеньмістким радикалом. У цьому відношенні радикально-ланцюгове окиснення відрізняється порівняно високою селективністю, що залежить від малої активності пероксидних радикалів. Реакційна здатність атомів водню у третинного, вторинного і первинного атомів вуглецю відноситься приблизно як 100:10:1.
Рівнобіжне утворення речовин з різними функціональними групами (утворення спиртів і кетонов з вуглеводнів, карбонових кислот і ангідридів з альдегідів) можна регулювати, підбираючи відповідні параметри процесу.
При послідовних перетвореннях, крім розглянутих раніше продуктів, можуть виходити оксикетони, дікетони, окси- і кетокарбонові кислоти та ін. Спостерігається і повне окиснення до СО2. Головним способом зниження ролі цих побічних реакцій є регулювання ступеня конверсії.
Усі процеси радикально-ланцюгового окиснення підрозділяються на дві групи:
1.Процеси, що протікають з цільовим одержанням речовин, стійких до подальшого окиснення (нижчі ароматичні та аліфатичні кислоти), коли ступінь конверсії не грає істотної ролі для селективности (ступінь конверсії a = 95-99 %), проміжну речовину можна повертати на окиснення разом з неперетвореним реагентом.
2.Процеси, спрямовані на цільовий синтез проміжних речовин, схильних до подальшого розкладання або окиснення (одержання гідропероксидів, спиртів, кетонів, вищих карбонових кислот), коли ступінь конверсії відіграє важливу роль і її обмежують величиною 5-30 %. У цьому випадку істотні витрати на регенерацію і рециркуляцію неперетвореного реагенту, ці процеси потрібно оптимізувати з урахуванням змін селективності і ступеня конверсії.
Великий вплив на селективність робить температура. Енергія активації побічних реакцій звичайно вище, тому роль останніх росте зі збільшенням температури, а селективність падає. У результаті кожен процес має деяку оптимальну температуру. Підвищення температури може відігравати роль у перекладі процесу в дифузійну або близьку до неї область протікання реакції: процес відбувається в прикордонній плівці, проміжні продукти не встигають продифундувати в об’єм рідини і переокиснюються. Тому важливу роль грає ефективна турбулізація реакційної суміші при барботируванні газу-окиснювача, що сприяє переходу процесу в кінетичну область, розвитку поверхні контакту фаз і інтенсифікації процесу. Таким чином, вибір оптимальних умов процесу є досить складним.
окиснення вуглеводень кинетика каталіз
4. Реактори процесів рідинофазного окиснення
Велике число процесів окиснення здійснюють у рідкій фазі шляхом барботирування повітря (рідше - технічного кисню) через вихідний органічний реагент, у якому поступово накопичуються продукти реакції. Якщо вибір температури залежить від інтенсивності і селективності процесу, то тиск підбирається, головним чином, з метою підтримки реакційної маси в рідкому стані.
Найбільше застосування одержали барботажні колони висотою 10-15 м і діаметром 2-3 м. Їх у деяких випадках секціонують горизонтальними сітчатими або ковпачковими тарілками або з'єднують у каскади. Оскільки карбонові кислоти кородують звичайну сталь, для виготовлення апаратури застосовують алюміній, титан або деякі леговані сталі, стабільні до дії органічних кислот.
Важливою обставиною є спосіб відводу великої кількості виділенного тепла. Маються системи з внутрішніми теплообмінниками, що ускладнює конструкцію реактору. Більш кращі реактори з выносними теплообмінниками і циркуляцією рідини через них. Ще вигідніше відводити тепло за рахунок випару вихідного вуглеводню або розчинника, які конденсують з відходячого газу у зворотному конденсаторі і повертають у реактор. У нових установках, що працюють при температурі вище 1500С, за рахунок реакційного тепла виробляють пар, а тиск використовують для часткового поділу суміші, для одержання холоду і т.д.
Схеми реакторів для проведення процесів рідинофазного окиснення представлені на рис. 1.
4.1 Характеристика реакторів
1. Реактор періодичної дії з виносним охолодженням за рахунок циркуляції рідини через водяний холодильник. Циркуляцію можна здійснювати й у протилежному напрямку, у тому числі і природним шляхом – за рахунок розходження щільностей рідин у колоні та у циркуляційному контурі. Вихідний реагент завантажують в апарат по закінченні попередньої операції, підігрівають до потрібної температури (у цей час замість води в холодильник надходить пара) і починають надавати повітря. Розподільним пристроєм служить перфорована труба, сітчата або ґратчаста тарілка.
2. Безперервне проведення процесу в одиничній барботажній колоні можливе при цільовому одержанні продуктів, стійких до подальшого окиснення (оцтова кислота, ароматичні кислоти). Окиснюєму речовину і повітря надають у низ реактора (прямоток), а продукти відбирають зверху. Відвід тепла можна здійснювати циркуляцією через водяний холодильник або вбудовані внутрішні холодильники.
3. Якщо селективність процесу залежить від ступеня конверсії вихідної речовини, то одинична барботажна колона не вигідна для безперервного процесу через сильне перемішування рідини. Такий процес краще проводити в каскадних барботажних колонах: рідка реакційна маса послідовно перетікає з колони в колону, а повітря надають роздільно в кожну. Теплоотвід може здійснюватися за рахунок випару вуглеводню або розчинника. Їх пари конденсуються в зворотних холодильниках, що знаходяться над кожною колоною.
4. Тарілчаста колона. У ній рідина перетікає зверху вниз з однієї тарілки на іншу, а повітря рухається протитоком. Для охолодження в змійовиках, поміщені в шари рідини на кожній тарілці, пропускають воду. Можливе і виносне охолодження.
В усіх реакторах періодичної і безперервної дії режим окиснення регулюють, змінюючи швидкість подачі окиснювача і вихідного органічного реагенту. Температуру звичайно замірять у декількох крапках по висоті реактора; реакційну масу періодично аналізують.
При рідинофазному окисненні необхідно виключити утворення вибухонебезпечних сумішей у місцях, де мається суцільна газова фаза, тобто у верхній частині барботажних колон і в просторі над кожною тарілкою (рис. 1, г). Це досягається високим ступенем конверсії кисню в сукупності з вибором тиску в залежності від летучості вихідної органічної речовини. Іноді в простір над рідиною передбачена подача азоту і т.д.
5. Окиснення вуглеводнів у гідропероксиди
Гідропероксиди застосовуються як проміжні продукти (наприклад, у виробництві фенолу та ацетону), ініціатори полімеризації, у процесах епоксидирування олефінів:
![]()
C6H5-CH(CH3)2![]() C6H5-C-(CH3)2
C6H5-C-(CH3)2
![]() C6H5OH
+ CH3-CO-CH3
C6H5OH
+ CH3-CO-CH3
![]()
RH![]() ROOH
ROOH ![]() ROH + CH3-CH-CH2O
ROH + CH3-CH-CH2O
5.2 Отримання гідропероксидів
У промисловості в найбільш великих масштабах одержують гідропероксид ізопропілбензолу (кумолу), у менш значних – гідропероксиди мета- і парацимолу (ізопропілтолуолу) і мета- і парадіізопропілбензолу для їх наступного перетворення у фенол, мета- і пара-крезол, резорцин, гідрохінон. Для епоксидирування олефинів використовують, головним чином, гідропероксиди етилбензолу та ізобутану. Усі вони є відносно стабільними речовинами. При одержанні концентрованих гідропероксидів (80-95 %) потрібні спеціальні міри безпеки: відсутність перегрівів і каталізаторів розкладання – металів перемінної валентності та їх солей, кислот.
При окисненні вуглеводнів гідропероксди утворюються за радикально-ланцюговим механізмом. Інгібітори (фенол, олефіни, сіркомісткі з'єднання) сильно гальмують процес, тому вихідні вуглеводні повинні бути ретельно очищені від небажаних домішок.
Ізопропілбензол, отриманий алкілуванням у присутності твердого фосфорнокислого каталізатора, не придатний для окиснення. Для зменшення індукційного періоду у вихідну сировину додають гідропероксид. Солі металів перемінної валентності розкладають гідропероксиди, однак в окремих випадках їх невеликі добавки прискорюють реакцію. Такий же ефект робить мідь, навіть якщо вона присутня в складі металу, який йде на виготовлення апаратури.
При одержанні гідропероксидів завжди утворюються побічні продукти: спирти, кетони, діметилфенілкарбінол, ацетофенон, моно- і дигідропероксиди.
Підвищенню селективності сприяє зниження температури і ступеня конверсії, параметри підтримують на оптимальному рівні, що залежить від економічних факторів. Температура складає 100-1500С, корисно знижувати її по мірі нагромадження гідропероксиду, щоб сповільнити його розкладання. Ступінь конверсії складає від 30 до 10 %.
Для одержання алкілароматичних гідропероксидів використовують реактори тарілчастого типу (рис. 1, г) або каскад окисних колон (рис. 1, в). Окислення ведуть повітрям при тиску 0,3-0,5 МПа для ізопропілбензолу і 5-8 МПа – для ізобутану (в останньому випадку тиск необхідний для підтримки суміші в рідкому стані).
Отриманий розчин гідропероксиду і побічних продуктів у вихідному вуглеводні звичайно “зміцнюють” або концентрують шляхом відгону вуглеводню.
5.3 Кислотне розкладання гідропероксидів
Гідропероксиди здатні до розпаду під впливом кислотних і лужних каталізаторів. У присутності вже невеликої кількості сильної кислоти (наприклад, 0,1 % H2SO4) гідропероксиди розпадаються з утворенням фенолів і карбонільних з'єднань. Виходить невелика кількість смол складної будівлі. При підвищенні концентрації кислоти і температури стає можливим перетворення ацетофенону та ацетону в окис мезитилу:
![]() 2СН3-СО-СН3
2СН3-СО-СН3![]() СН3-СО-СН2-С(СН3)2
СН3-СО-СН2-С(СН3)2![]() СН3-СО-СН=С(СН3)2
СН3-СО-СН=С(СН3)2
окис мезитилу
ОН
Розкладання гідропероксидів характеризується високою швидкістю: практично повне перетворення, у присутності 0,05-0,1 %-вої сірчаної кислоти при температурі 50-600С, досягається за 2-3 хвилини. Реакція гальмується водою і прискорюється утворюємим фенолом.
Через високу швидкість процесу при його промисловій реалізації дуже важливим є ефективний відвід великої кількості тепла, що виділяється, (Q=2080 кДж/кг). Для цієї мети застосовують розріджувачі, якими є продукти реакції або ацетон.
Для проведення реакції застосовують проточно-циркуляційні установки, коли виділене тепло знімають у трубчастому реакторі за рахунок охолодження його водою. Реакційну суміш на виході з реактора частково відводять на подальшу переробку, але основну кількість направляють на рециркуляцію: додають кислоту, каталізатор і в насосі змішують з вихідним гідропероксидом. За такою системою час контакту лімітується тепловідводом і є завищеним, рециркуляція веде до підвищеного виходу побічних речовин (на 1 т фенолу виходить 100-150 кг відходів).
Інший спосіб складається в проведенні реакції в розчині ацетону і відводі тепла за рахунок його випару. Ацетон конденсують у зворотному холодильнику і повертають до реактору, який можна секціонувати поперечними перегородками. Це, поряд зі зменшенням концентрації фенолу в розчині і часі контакту, знижує вихід побічних продуктів.
Кислотним розкладанням гідропероксидів одержують:
- фенол (С6Н5ОН) – проміжний продукт у виробництві барвників, лікарських і вибухових речовин;
- гідрохінон, резорцин – застосовують для одержання легко відтверджуємих фенолальдегідних полімерів, інгібіторів;
- b-нафтол – використовують у виробництві барвників;
- ацетон – прекрасний розчинник.
Технологічна схема кумольного методу одержання фенолу та ацетону представлена на рис. 2.
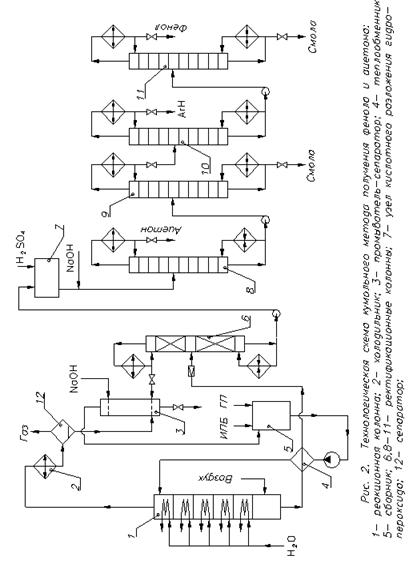
Рис. 2. Технологічна схема кумольного методу отримання фенолу та ацетону:
1 – реакційна колона; 2 – холодильник; 3 –промивач-сепаратор; 4 – теплообмінник; 5 – збірник; 6, 8–11 – ректифікаційні колони; 7 – вузол кислотного розкладення гідропероксиду; 12 – сепаратор.
6. Окиснення парафінів
Існують три напрямки окисної переробки парафинів:
1.Окиснення в газовій фазі, для одержання нижчих спиртів і альдегідів.
2.Термічне окиснення в рідкій фазі в присутності борної кислоти для синтезу вищих вторинних спиртів.
3.Каталітичне окиснення в рідкій фазі для одержання карбонових кислот.
Процес каталітичного окиснення в рідкій фазі має найбільше практичне застосування.
6.1 Окиснення нижчих парафінів у газовій фазі
Здатність нижчих парафінів до окиснення залежить від довжини ланцюга. Так, у відсутності каталізаторів і при звичайному тиску метан починає окиснюватися при температурі 4200С, етан – при 2850С, пропан – при 2700С. З підвищенням тиску початкова температура окиснення знижується (наприклад, метан при тиску 10 МПа реагує з киснем уже при температурі 3300С). Гомогенні ініціатори (оксид азоту, бромоводень HBr), а також гетерогенні каталізатори дозволяють прискорити процес і здійснити його при більш низькій температурі.
Окиснення в газовій фазі може відбуватися зі збереженням або деструкцією вуглецевого ланцюга. Пряме окиснення метану у формальдегід утрудняється відносною легкістю подальшого окиснення і розкладання формальдегіду:
СН4 ![]() НСНО
НСНО ![]() НСООН
НСООН
![]() СО2
+ Н2О
СО2
+ Н2О
Тому задовільна селективність за формальдегідом досягається тільки при дуже малому ступені окиснення метану в умовах недоліку кисню, що можливо лише при великій кратності циркуляції вихідного вуглеводню. Спосіб виявився економічно не вигідним.
Газофазне окиснення парафинов С3-С4 дає суміш спиртів і карбо-нильних з'єднань, що утворилися зі збереженням і з деструкцією вуглецевого ланцюга:
![]() СН3-СН2-СН2ОН
СН3-СН2-СН2ОН![]() СН3-СН2-СНО
СН3-СН2-СНО
СН3-СН2-СН3![]() СН3-СНОН-СН3
СН3-СНОН-СН3 ![]() СН3-СО-СН3
СН3-СО-СН3
СН3ОН + СН3СНО
С2Н5ОН + НСНО
Кількість продуктів деструкції росте з підвищенням температури, складаючи, наприклад, для пропану 76 і 98 %, відповідно при температурі 250-3730С. Даний процес реалізований тільки в США і має задачею одержання формальдегіду, ацетальдегіду, метанолу і так званого змішаного розчинника, що містить спирти С2-С3, ацетон, метилетилкетон. Окиснення парафинів С3-С4 ведуть при температурі 4000С и недоліку кисню в пустотілому адіабатичному реакторі під тиском 0,7-2,0 МПа. Недолік процесу – складність одержуваної суміші, що викликає підвищені капітальні та й енергетичні витрати на стадії поділу.
7. Гетерогенно-каталітичне окиснення вуглеводнів та їх похідних
Гетерогенно-каталітичне окиснення придбало велике значення при здійсненні ряду процесів, які не можна успішно реалізувати за допомогою радикально-ланцюгових реакцій окиснення. Серед них найважливішими є наступні:
1.Окиснення парафинів та їх похідних за насиченим атомом вуглецю зі збереженням подвійного зв'язку:
СН2=СН-СН3 + О2 ® СН2=СН-СНО + Н2О
2.Окисний амоноліз олефінів та інших вуглеводнів з одержанням нітрилів:
RCH3 + NH3 + 1,5O2 ® RCN + 3Н2О
3.Окиснення ароматичних та інших вуглеводнів з утворенням внутрішніх ангідридів ди- або тетракарбоновых кислот:
![]() СО
СО
![]()
![]() НС
НС
![]() + 4,5О2 ® çïО + 2CO2 + H2O
+ 4,5О2 ® çïО + 2CO2 + H2O
![]() НС
НС
СО
4.Прямий синтез етиленоксиду:
![]()
СН2=СН2 + 0,5О2 ® СН2-СН2О
Усі ці процеси мають дуже велике практичне значення, тому що одержані продукти широко використовуються як мономери (акрилова, метакрилова кислоти, акрилонітрил, малеїновий і фталевий ангідрид) і проміжних продуктів для синтезу пластифікаторів, розчинників та інших мономерів (фталевий ангідрид, етиленоксид, нітрили, акролеїн).
7.1 Теоретичні основи процесу
7.1.1 Гетерогенні каталізатори окиснення і механізм реакцій
Застосовуються наступні каталізатори:
1.Метали міді і срібла, з яких більш легко окиснюєма мідь функціонує у виді оксидів, які утворюються в поверхневому шарі. Інші метали (платина, паладій) приводять до повного окиснення до СО2 і води.
2.Оксиди перехідних металів Cu+Cu2O, V2O5, інші оксиди неактивні або сприяють повному окисненню.
3.Суміші оксидів і солі перехідних металів, особливо ванадати, станнати, вольфрамати, молібдати Zn, Co, Bi (ZnO×V2O5, CoO×WO3, Bi3O3×MoO3), але ферріти і хроміти викликають повне окиснення.
Каталізатор застосовують у виді стружок, сіток (Cu), зерен (V2O5) або нанесеними на пористий носій (Ag, Cu, солі), нерідко з добавками різних промоторів.
У механізмі гетерогенних реакцій окиснення важливу роль грає адсорбція реагентів на поверхні контакту. На металах кисень сорбируется дуже швидко, з наступним більш повільним проникненням у приповерхній шар. Неблагородні метали в результаті дають оксиди, а для срібла процес обмежується хемосорбцією з глибокою зміною властивостей приповерхневого шару. Кисень сорбується на контакті без дисоціації або з дисоціацією молекули, причому метал поставляє необхідні електрони і переводить адсорбуємий кисень у стан іон-радикалу:
Ag + O2 ® ![]()
![]()
![]()
Подібна хемосорбція кисню здійснюється на оксидних і сольових каталізаторах.
Вуглеводні сорбуюються на металах порівняно слабко і оборотно. Мічніше вони сорбуюються на оксидних і сольових каталізаторах:
М(n+1)+ + CH2=CH-CH3 « Mn+-CH2-![]() -CH3
-CH3
Маються два головних типи механізму гетерогенно-каталітичного окиснення. В одному з них вуглець сорбується на окисненій поверхні каталізатору, спочатку сорбуясь по іон-радикалу кисню, а потім, взаємодіючи з ним, з утворенням продуктів окиснення. Типовим прикладом є синтез етиленоксиду:
![]()
![]() + CH2=CH2 « AgOOCH2-
+ CH2=CH2 « AgOOCH2-![]() ®
® ![]() + CH2-CH2O
+ CH2-CH2O
Той же механізм при окисненні бензолу в малеїновий ангідрид, що йде через проміжне утворення хінону.
![]() ОО· +®
ОО· +® ![]() ОО ¾
ОО ¾ ![]()
![]() ОО ¾¾
ОО· ®
ОО ¾¾
ОО· ®
СО
НС
![]() О == О
О == О ![]() çï
çï
СО
Інший розповсюджений механізм – окиснювально-відновлюваль-ний. На іоні металу сорбуєтья вуглець і він окиснюється киснем решітки каталізатору, метал при цьому відновлюється в нижчий валентний стан і потім знову взаємодіє з киснем, переходить у початкову форму:
2КО + СН2=СН-СН3 ® 2К + СН2=СН-СНО + Н2О
2К + О2 ® 2КО
Цей механізм характерний для окиснення олефінів і метилбензолів. Він підтверджується тим, що очікувані продукти можуть виходити на каталізаторі під час відсутності кисню, а стадії окиснення вуглеводню та окиснення каталізатору можна проводити роздільно. Однак точно представити їх картину не можливо, тому що ще неясні будівля поверхневих продуктів та їх роль в окисненні.
7.1.2 Кінетика і селективність гетерогенно-каталітичного окиснення
Загальним у кінетиці окиснення є гальмуючий вплив продуктів окиснення, які адсорбуються на поверхні сильніше, ніж вихідні вуглеводні. Енергія активації при гетерогенному окисненні олефінів складає 63-84 кДж/моль, а для ароматичних з'єднань – 105 кДж/моль.
Схема реакцій з рівнобіжним і послідовним утворенням побічних продуктів повного окиснення :
![]() RH
RH ![]() цільовий продукт
цільовий продукт ![]() СО2 + Н2О
СО2 + Н2О
+О2
Придушення рівнобіжних реакцій повного окиснення за рахунок варіювання співвідношення реагентів звичайно виявляється неможливим, але зате в цьому відношенні значну роль грає температура. Енергія активації повного окиснення на 21-42 кДж/моль вище, ніж для цільового процесу, селективність росте зі зниженням температури, для етилену. З урахуванням одержання досить високої продуктивності, існує оптимальна область температур, обумовлена економічними розуміннями. З цього ж ясні неприпустимість місцевих перегрівів і важлива роль тепловідводу, який збільшується високою екзотермічністю процесів окиснення. Через послідовне окиснення цільової речовини в продукти повного окиснення селективність часто падає зі збільшенням ступеню конверсії вуглеводню. З урахуванням витрат на регенерацію непрореагувавшого вуглеводню в кожнім процесі існує деякий оптимальний ступінь конверсії, обумовлений економічними факторами. За інших рівних умов її регулюють двома способами: часом контакту або застосуванням недоліку кисню.
Нарешті, найважливішу роль грає і сам каталізатор, спосіб його готування. Додавання різних модифікаторів або застосування сумішей оксидів і солей здатно сильно змінювати активність і селективність контакту. Деякі каталітичні отрути (галогени, селенів), дезактивуючи каталізатор (срібло) окиснення етилену, істотно підвищують його селективність. Оксиди молібдену і вісмуту, які в індивідуальному виді ухвалюють повне окиснення олефінів, у формі молібдату Bi (Bi2O3:Mo3 = 1:2) є селективними каталізаторами гетерогенного окиснення пропілена. Великий вплив має носій, розмір зерен каталізатора, його пористість і т.д. Через можливість послідовного окиснення цільової речовини і високу швидкість самої хімічної реакції на поверхні каталізатора і перехід процесу у внутрідифузійну область досить не бажаний, тому використовують каталізатор з невеликими зернами і порівняно великими порами.
7.2 Реактори процесів гетерогенно-каталітичного окиснення
Реакції гетерогенно-каталітичного окиснення часто проводять при атмосферному тиску. Але з'являється усе більше виробництв, де ці реакції ведуть під тиском від 0,3 до 2,0 МПа, що дозволяє інтенсифікувати процес, знизити габарити апаратури і полегшити виділення неперетвореного вуглеводню і продуктів. Температура для різних процесів змінюється від 230-3000С до 400-5000С. Через вибухонебезпечність сумішей вуглеводнів з киснем мається кілька способів виходу за межі небезпечних концентрацій: окиснення рециркулюючими газами з добавкою свіжого повітря і кисню при низькій концентрації олефінів (3-5 % об.), окиснення надлишку вуглецю невеликою кількістю технічного кисню, розведення суміші водяною парою.
Одним з основних питань при конструюванні реакційних апаратів для газофазного окиснення є відвід тепла і виключення зон перегріву. Через невеликі коефіцієнти тепловіддачі від газу до стінки ця задача є більш складною, чим при рідинофазних процесах окиснення. Усе більшого значення набуває і проблема підвищення теплового коефіцієнта корисної дії установок, що зводиться до утилізації реакційного тепла для виробництва водяної пари.
Через високу екзотермічність окиснення адіабатичні реактори не знайшли застосування в цьому процесі. Розповсюджений трубчастий реактор зі стаціонарним шаром каталізатору та охолоджуваним хладоагентом (рис.3, а). Труби мають діаметр 10-25 мм, що сприяє відводу тепла і встановленню більш рівномірної температури по діаметру. Щоб краще використовувати катализаторний об’єм, в апарат надають реагенти попередньо підігрітими. Найкращий спосіб відводу виділеного тепла – це випар у міжтрубному просторі водяного конденсату, що генерує водяну пару того або іншого тиску в залежності від температури реакції. Іноді використовують охолодження стороннім теплоносієм (розплави солей, даутерм), що у свою чергу охолоджується киплячим водним конденсатом, що дає технологічну пару. Перевагою трубчастих контактних апаратів є простота їх пристрою та обслуговування, а також близькість до моделі ідеального витиснення, що сприяє підвищенню селективності; недоліки – нерівномірність температури по шару каталізатора, мала частка корисного обсягу, велика витрата металу.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
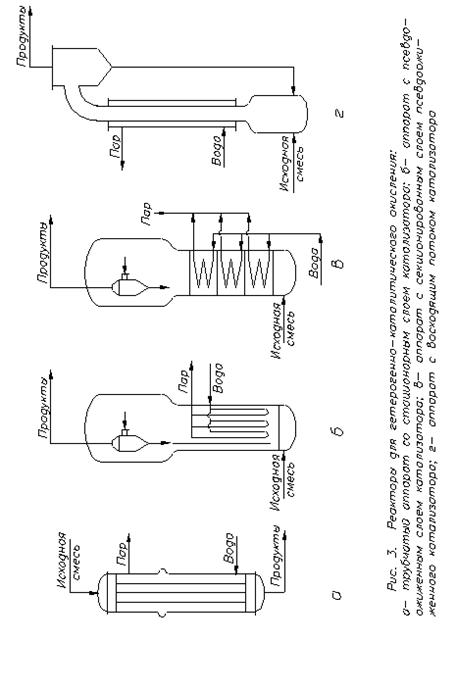
Рис. 3. Реактори для гетерогенно-каталітичного окиснення:
а – трубчастий реактор зі стаціонарним шаром каталізатору; б – апарат з псевдозрідженим шаром каталізатору;
Для багатьох процесів окиснення використовують реактори з псевдозрідженим шаром гетерогенного каталізатору (рис.3, б). Вони подібні реакторам каталітичного крекінгу, але не мають регенераторів, тому що при окисненні утворення смол і сажі незначно, каталізатор зберігає активність протягом багатьох місяців і навіть декількох років. Реагенти можна надавати холодними, каталізатор використовують у зміцненій мікросферичній формі, який знаходиться у постійному витанні.
Реактор звичайно облагоджений розподільними ґратами з охолодними трубами, у яких регенерується пара, і циклонами для уловлювання часток, віднесених газом. У такому апараті ідеально вирішується проблема тепловідводу і підтримки рівномірної температури, але відбувається зворотне перемішування, що знижує селективність процесу. Для усунення цього недоліку використовують секціоновані апарати, у яких крім основних розподільних ґрат мається кілька ґрат, що поділяють основний об’єм на секції (рис.3, в). Зменшення зворотного перемішування досягається також у реакторах з висхідним потоком каталізатора (мал.3, г), що переміщається нагору разом з газом. Реакційна труба прохолоджується рубакою з киплячим водним конденсатом. Каталізатор відокремлюється в сепараторі або циклоні і повертається по трубі в нижню частину реактора, яка називається дозатором. У цьому апараті умови теплопередачі гірші, ніж у попередніх, тому що зовнішнє охолодження, при досить широкій реакційній трубі, менш ефективно. Такий реактор можна виконати у виді багатотрубного агрегату, охолоджуваного через міжтрубний простір.
При виборі матеріалу апаратури для газофазного окиснення варто враховувати корозію не тільки карбоновими кислотами, але і сумішами діоксиду вуглецю з водяною парою, також можуть каталізуватися небажані процеси повного окиснення. У зв'язку з цим ці апарати виконують з легованих сталей.
7.3 Окиснення олефінів по насиченому атому вуглецю
Цим методом одержують акролеин і акрилову кислоту:
СН2=СН-СН3![]() СН2=СН-СНО
СН2=СН-СНО![]() СН2=СН-СООН
СН2=СН-СООН
Акролеїн – рідина з різким запахом, добре розчинна у воді та утворююча з нею азеатропну суміш. При тривалому збереженні і нагріванні легко полімеризується в циклічні та лінійні полімери, для його збереження використовують інгібуючи добавки. Застосовується для одержання акрилової кислоти та її ефірів, алілового спирту, гліцерину.
7.3.1 Окиснення пропілену в акролеїн
Процес супроводжується утворенням побічних речовин: ацетальдегіду, ацетону, оцтової кислоти, акрилової кислоти, СО2, СО.
Каталізатори – Cu2O 0,1-0,2 % на пемзі, карборунді або Al2O3, або навіть мідні трубки реактору; Bi2O3×MoO3; Bi2O3×MoO3×Р2О5, утримуючі промотори (оксиди телуру і міді). На цих каталізаторах досягається високий ступінь конверсії при малому часі контакту і помірній температурі. На оксиді міді t - 0,2 сек., Т – 370-4000С або t - 0,2 сек., Т – 320-3500С. На молібдатах застосовують більш високую температуру - 400-5000С, t - 1-2 с, тиск від 0,1 до 1,0 МПа.
Склад вихідної суміші обмежується межами висадження, тому додають водяну пару (25(50 %), що також сприяє селективності за рахунок десорбції акролеїну. Як газ-окиснювач використовують технічний кисень або повітря. Останній дешевше технічного кисню, але розбавляє реакційні гази та утрудняє виділення і рециркуляцію речовин. Співвідношення пропилену та окиснювача різне: 42-44 % об. С3Н6, 8-10 % об. О2, 46-50 % об. Н2О – надлишок пропілену; 7-8 % об. С3Н6, 67 % об. повітря, 25 % об. Н2О – надлишок окиснювача. У першому випадку необхідна рециркуляція неперетвореного пропілену, чим і порозумівається застосування не повітря, а технічного кисню; ступінь конверсії складає від 60 до 100 %, селективність – 70-90 %.
Реакцію проводять у різних реакторах, але частіше у кожухотрубчастих реакторах зі стаціонарним шаром, охолоджуваних розплавом солей. Розплав циркулює через котел-утилізатор, генеруючи пару високого тиску. Реакційні гази проходять потім абсорбер, де продукти окиснення поглинаються водою і виходить 1,5-2,0 % розчин акролеїну, що містить ацетальдегід, ацетон і невелику кількість пропіонового альдегіду. Ацетальдегід легко відокремлюється ректифікацією, а для очищення акролеїну від близькокиплячого пропіонового альдегіду (температура кипіння складає 490С) використовують екстрактивну дистиляцію водою. Отриманий акролеїн містить 99 % основної речовини з домішкою води і пропионового альдегіду.
7.3.2 Одержання акрилової кислоти
Для одержання акрилової кислоти використовується вісмут-молібденовий каталізатор з різними промоторами (Те, Со, Р та ін.), але умови реакції більш м'які: Т = 200-3000С, t = 0,5-2,0 сек. Побічно утвориться оцтова кислота, СО2 і СО, селективність складає 90 %.
Для реалізації процесу одержання акрилової кислоти в промисловості комбінують окиснення пропілену в акролеїн і окиснення акролеїну в акрилову кислоту. На початку здійснили одностадійний процес прямого окиснення пропілену в акрилову кислоту в одному реакторі. Однак вихід її склав всего 70 %, оскільки сполучення двох стадій з різними для них оптимальними умовами не могло сприяти росту селективності.
Зараз застосовується двостадійний процес одержання акрилової кислоти (рис. 4). У реактор першої стадії надають суміш, що містить 4-7 % пропілену, 50-70 % повітря, 25-40 % водяної пари; температура складає 300-4000С; каталізатор - Bi2O3×MoO3. У реакторі другої ступені температура складає 250-3000С, використовуваний каталізатор - Bi2O3×MoO3. Отриманий водний розчин містить 20-30 % акрилової кислоти з домішкою оцтової кислоти. Для виділення цільового продукту застосовують екстракцію низкокиплячим органічним розчинником. Його відганяють з екстракту і повертають на витяг, а при ректифікації залишку одержують акрилову та оцтову кислоту. Вихід складає 80-85 % за пропіленом.
7.4 Окисний амоноліз вуглеводнів
Вуглеводні здатні взаємодіяти з аміаком при високій температурі, утворюючи нітрили:
RCH3 + NH3 ® RCN + 3H2
Крім того, нітрили утворяться з альдегідів і аміаку через проміжну стадію дегідрування амінів:
RCHО + NH3 ![]() RCH=NH
RCH=NH ![]() RCN
RCN
Ці два процеси сполучили з окисненням – це процес окисного амонолізу вуглеводнів:
RCH3 + NH3 + 1,5О2 ® RCN + 3H2О
Вперше окисному амонолізу піддали метани, а потім олефіни і метилбензоли.
![]()
![]()
![]()
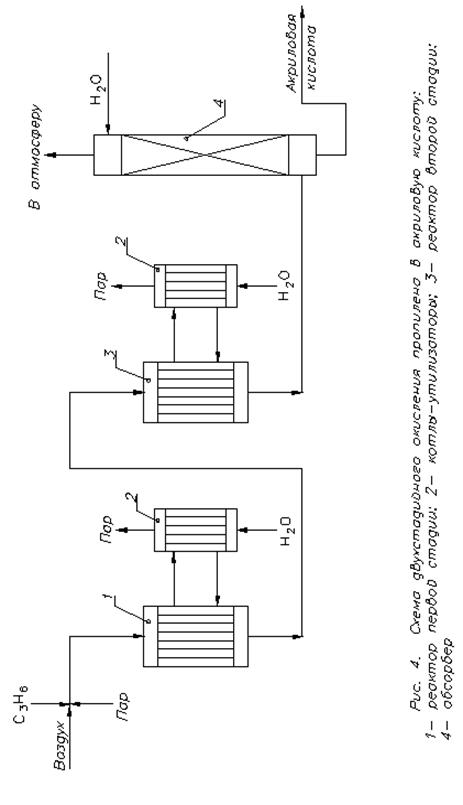
Рис. 4. Схема двохстадійного окиснення пропілену в акрілову кислоту: 1 – реактор першої стадії; 2 – котли-утилізатори; 3 –реактор другої стадії; 4 – абсорбер.
Література
1. Лебедев Н.Н. Химия и технология основного органического и нефтехимического синтеза. – М.: Химия, 1988. С. 338-367.
2. Тимофеев В.С., Серафимов Л.А. Принципы технологии основного органического и нефтехимического синтеза. – М.: Высшая школа, 2003. С. 16-22, 335-348.
| Обследва процеса на реформинг на природния газ и получаване на водород | |
|
Министерство на образованието и науката Университет "Професор доктор Асен Златаров" Факултет "Технически науки" гр. Бургас ДИПЛОМНА РАБОТА на Петър ... Катали-затор съдържащ ѭ- Al2O3 може да бъде нагряван с водни пари до 800 0С без да се образуват шпинели. Обемната скорост зависи от налягането на процеса и използвания катали-затор и се колебае в границите от 1000 - 1500 ч-1, при атмосферно налягане до 6000 - 8000 ч-1 при 2Mpa. |
Раздел: Промышленность, производство Тип: дипломная работа |
| Экономическая система | |
|
1. Эк-я с-ма - ед-во ч-ка и общ-го пр-ва В любой ЭС существует 2 вида отношений: 1-отношение людей к природе; 2. отношение людей друг к другу. В ... Тпр. ц = Кк/Тсм* S(Тт. ц+ Тк+ Ттр + Тм. о) + Тест/24., где: Преим-ва: созд-ся благопр-е усл-я для внедр-я новой техники, поточного метода орг-ции пр-ва, механизации и автом-ции, сокращ-ся дл-сть цикла. |
Раздел: Рефераты по экономике Тип: шпаргалка |
| Разработка отварочной технологии производства пива | |
|
ВВЕДЕНИЕ На сегодняшний день в России складывается ситуация, при которой основную долю рынка пива занимают производители-гиганты. В то же время в ... Так, использование 30%-ной кукурузной крупки в заторе приводит к снижению количества ѭ-аминного азота на 30%. ѭ-горькие кислоты (гумулоны). |
Раздел: Промышленность, производство Тип: дипломная работа |
| Проектирование систем электроснабжения промышленных предприятий на ... | |
|
Введение Передача электроэнергии от источников к потребителям производится энергетическими системами, объединяющими несколько электростанций ... Qр, он = Рр, он - tg ѭ, где tg ѭ соответствует cos ѭ осветительной установки. Катал. |
Раздел: Промышленность, производство Тип: дипломная работа |
| Теоретичні основи теплотехніки | |
|
1. Характеристика курсу Дистанційний курс "Термодинаміка та теплотехніка " Загальна кількість кредитів: національних 2 ECTS 3,5 Лекційне навантаження ... Ексергія речовини є максимальна робота, яку може виконати робоче тіло в оборотному процесі з навколишнім середовищем в якості джерела дармової теплоти, якщо в кінці ц"ого процесу ... Кількі сть теплоти, яка відводиться від 1 кг ідеального газу в процесі його стисненняв циліндрі компресора |
Раздел: Промышленность, производство Тип: учебное пособие |